Airing Out an Antioxidant Role for the Tumor Suppressor p53
One might predict that after two decades of intense scrutiny, new functions for the most extensively characterized tumor suppressor are unlikely to be identified. The recent work by Sablina and coworkers (1) suggests otherwise. We know p53 largely exerts its tumor suppressor activity by preventing damaged DNA from being transmitted to daughter cells by orchestrating the expression of genes involved in cell cycle progression, DNA repair, and both intrinsic and extrinsic apoptotic pathways (2). Whether increased p53 forces a cell to arrest or to engage in apoptosis depends on the tissue of origin, the nature of the DNA damage and, finally, the extent of the DNA damage. p53 is reported to promote apoptosis through transcription-dependent and -independent pathways (3). Of the two pathways, transcriptional regulation of pro- and anti-apoptotic genes by p53 is most well-accepted and characterized.
All mammalian cells encounter DNA damage either through exogenous factors (such as UV light, ionizing radiation, and chemotherapy) or endogenous factors [such as spontaneous deamination of bases or oxidative base modification by reactive oxygen species (ROS)]. ROS are normal byproducts of cellular processes such as respiration, cell injury, metabolic hydroxylation, inflammation and phagocytosis. It is estimated that each day ROS modify 20,000 bases of DNA in each of our cells (1, 4). Consequently, valuable cellular antioxidant mechanisms have evolved to defend against this seemingly massive source of DNA damage. These include repair processes, detoxifying enzymes such as catalases, peroxidases, transferases, reductases, superoxide dismutases, and small protein or peptide thiols, such as metallothionein or glutathione (5, 6). Links between ROS and p53 activity have previously been reported (7, 8). Namely, activated p53 increases cellular ROS by enhancing the transcription of proapoptotic genes (7, 8) (Figure 1⇓). Moreover, scavenging of ROS by antioxidant therapy decreases apoptosis induced by p53. Paradoxically, p53 also appears to regulate the expression of some well-known antioxidant genes, including glutathione peroxidase, mitochondrial superoxide dismutase 2, and mammalian sestrin homologs SESN1 and SESN2, which are involved in regeneration of overoxidized peroxiredoxins (9–12) (Figure 1⇓). These data beg the question: is p53 a prooxidant or an antioxidant? Or could it be both? In an attempt to decipher this apparent conundrum, Sablina et al. (1) used several molecular biological approaches and the old-fashioned pharmacological concept of dose-response to probe the role of p53 in regulating cellular ROS levels. They concluded that the role of p53 is contextually controlled: low amounts of p53 suppress ROS under normal physiological conditions but high p53 expression promotes ROS accumulation in conditions of profound pathological stress.
When a number of normal and cancer cell lines were depleted of p53 by small interfering RNA (siRNA), the authors found increased basal levels of ROS comparable in magnitude to that observed with the treatment of hydrogen peroxide. These results were corroborated using other techniques to reduce the expression of p53, such as the introduction of the p53 inhibitors HPV18-E6 or MDM2. Overexpression of a dominant negative mutant of p53 also increase ROS levels.
Using genetic approaches, the authors investigated how loss of p53 might contribute to increased ROS production. Depletion of p53 caused a significant decrease in the expression of antioxidant genes SESN2, GPX1 (encoding glutathione peroxidase-1), and the p53-inducible transcript T2 of SESN1 (12, 13), whereas the expression of pro-oxidant genes such as BAX, TP53I3 (also known as p53-inducible gene 3, PIG3), and BBC3 (which encodes Puma) remained undetectable in unstressed cells. The expression of SESN2, SESN1, and GPX1 was reduced in the organs of p53-null mice, although there were tissue-specific differences in the expression of these genes. Conversely, p53 induction by genetic restoration in p53-negative cells decreased ROS and induced antioxidant genes. As p53 concentrations reached supraphysiological levels (which is observed under severe cellular stress), p53 increased ROS production coincident with increases in the pro-oxidant genes TP53I3, BAX, and BBC3.
These results, supplemented with observations using two doses of hydrogen peroxide, led the authors to conclude that in response to nontoxic or “physiological” stress, p53 increases the expression of antioxidant genes, whereas in the gravely damaged cells, p53 increases the expression of prooxidant genes that further contributed to p53-induced apoptosis. The p53-dependent increase in ROS appeared to originate from mitochondrial leakage during apoptosis. Overall, these results emphasize the critical role that p53 has in determining cellular redox status.
Because half or more cancers have inactivating p53 mutations (14), a logical hypothesis would be that increased ROS production arising from loss of p53 activity could contribute to DNA oxidation and mutagenesis, and thus accelerate the development of cancer. The authors directly examined this hypothesis by monitoring the rate of formation of 8-oxoguanine (8-oxo-dG), the major product of DNA oxidation, and a major source of mutations. In p53-depleted cells and splenocytes from p53−/− mice, there was a significant increase in 8-oxo-dG as compared to the amounts of 8-oxo-dG found in control cells. This effect was attenuated by overexpression of the antioxidant genes SESN1 and/or SESN2. As might be predicted, elevated DNA oxidation increased mutation frequencies within the hypoxanthine-guanine phosphoribosyltransferase (HPRT) gene locus, a long-used method to assess mutagenesis, and this was inhibited by treatment with N-acetyl cysteine (NAC) or by overexpression of both SESN1 and SESN2.
The increased rate of mutagenesis and DNA oxidation suggests that the antioxidant function of p53 may directly contribute to the prevention of tumor development. To test this hypothesis, Sablina et al. (1) injected p53-depleted cells into athymic mice and showed considerable increase in tumor volume compared to control cells in which p53 was not depleted. Interestingly, this effect was reversed by dietary NAC supplementation. Most strikingly, however, was that NAC supplementation of p53−/− mice from birth approximately halved the number of observed karyotype abnormalities and decreased cancer incidence by more than ninety percent at six months after birth, when most untreated p53-null mice have succumbed to neoplasia.
Given that supplementation of p53-null mice with NAC was largely able to prevent formation of tumors, the authors provide some pharmacological insights into potential therapeutic approaches for preneoplastic lesions with loss of p53 activity. Accordingly, the authors’ work implies that antioxidant therapy might also be used as prophylactic agents to prevent development of cancer in patients with germline mutations in p53. Alternatively, antioxidant therapy may afford sufficient time to treat a more developed tumor effectively before it becomes resistant to conventional chemotherapies.
Whereas many groups have reported proof-of-principle findings in which reactivation of p53 was detrimental to the tumor, the findings have often been difficult to translate to other tumor or cell types (15, 16). These difficulties presumably arise from the different and distinct modes of p53 deregulation in these cells. A logical deduction based on these cell-specific findings is that, because of the magnitude of distinct p53 mutations in different cancers, a common small molecule solution to p53 reactivation is practically impossible. However, the fact that the antioxidant function of p53-mediated tumor suppression could be so easily recapitulated by supplementation of diet is promising and demands further study. What is unclear from the work presented is whether NAC supplementation has any effect in animals with established tumors. The evolution of a tumor often involves acquisition of multiple mutations involving various pathways and aneuploidy leading to significant tumor-induced stress, in which case antioxidant therapy may yield limited or no efficacy. It is entirely possible that the effect of antioxidant supplementation may depend intimately on the stage and the specific “oncoprofile” of the tumor. Also, it remains unclear whether antioxidant supplementation will have any chemopreventive effect on p53-positive preneoplastic lesions. One could argue that it is unlikely antioxidant supplementation would be efficacious, considering that basal p53 activity appears sufficient to curtail spontaneous tumorigenesis induced by endogenous ROS.
In conclusion, the study by Sablina et al. (1) provides evidence of an antioxidant role of p53 that protects against oxidative DNA damage and genomic instability under normal conditions. The relative proapoptotic and antiapoptotic functions of p53 would appear to depend at least in part on the cellular p53 concentration (Figure 1⇓) if not other factors, such as p53 subcellular localization, phosphorylation status and protein partner interactions. The new paradigm provides ample evidence that new information can be obtained from even a well-studied protein like p53.
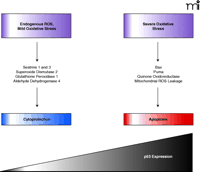
Model of concentration-dependent role of p53 in maintaining cell survival or causing apoptosis. Under normal cellular conditions, amounts of ROS are modest and cells experience only mild oxidative stress. In such situations, p53 positively regulates the expression of antiapoptotic genes that lead to cell protection. Under periods of extensive cellular stress, p53 concentrations increase and a new panel of genes are expressed that lead to apoptosis.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

John S. Lazo, PhD, (middle) is the Allegheny Foundation Professor of Pharmacology and the Director of the University of Pittsburgh Drug Discovery Institute. After graduating with a bachelor degree in Chemistry from the Johns Hopkins University, he completed his PhD in Pharmacology with Raymond Ruddon at the University of Michigan. He joined Alan Sartorelli as a postdoctoral fellow and remained on the faculty in the Department of Pharmacology at Yale University until 1987, when he became Chairman of Pharmacology at the University of Pittsburgh. His laboratory is currently interested in the biological role of protein phosphatases in normal and malignant cells and in the pharmacological actions of novel natural products. E-mail: lazo{at}pitt.edu; fax 412-648-9009.
Pallavi Bansal, BSc, MSc, (left) is a graduate student in John S. Lazo’s laboratory in the Department of Pharmacology at University of Pittsburgh. Her research focuses on understanding regulation of Cdc25 dual specific phosphatases following DNA damage.
Robert J. Tomko, Jr., BS, (right) is a pharmacology graduate student in John S. Lazo’s laboratory at the University of Pittsburgh. He is currently studying the role of oxidative and nitrosative post-translational modifications in the regulation of tyrosine phosphatase activity.

