Salvinorin A
From Natural Product to Human Therapeutics
Abstract
The hallucinogenic plant Salvia divinorum (i.e., “magic mint”) is a member of the Sage family that has been used for divination and shamanism by the Mazatecs. Over the past decade or so, S. divinorum has been increasingly used recreationally. The neoclerodane diterpene salvinorin A is the active component of S. divinorum, and recently, the κ opioid receptor (KOR) has been identified, in vitro and in vivo, as its molecular target. The discovery of KOR as the molecular target of salvinorin A has opened up many opportunities for drug discovery and drug development for a number of psychiatric and non-psychiatric disorders.

Introduction
Salvia divinorum is a perennial herb of the Lamiaceae (mint) family that is indigenous to the Sierra Mazateca of Oaxaca, Mexico (1, 2) (Figure 1⇓). Common names for the herb include ska Maria Pastora and la Maria, reflecting the Mazatec belief that Salvia is the incarnation of the Virgin Mary(1). S. divinorum has been traditionally used by Mazatec curanderos (i.e., folk healers) to produce hallucinogenic experiences essential for spiritual divination. S. divinorum has also been used in traditional healing practices for ailments as diverse as diarrhea, headache, and rheumatism, as well as the magical disease known as panzón de barrego (i.e., swollen belly), which is said to be caused by a curse from an evil sorcerer (1). S. divinorum was discovered by Wasson and Hofmann in 1962 and was subsequently described by Epling as a new species of Salvia (2).
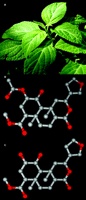
The magic mint and its products. A.Salvia divinorum is a member of the Lamiaceae (mint) family that has been traditionally used by Mazatec shamans for spiritual divination, and more recently S. divinorum has become increasingly used as a recreational hallucinogen. B. Salvinorin A is the active component of S. divinorum. C. Salvinorin B is a potential salvinorin A metabolite resulting from ester hydrolysis and is relatively inactive at KOR. Carbon atoms are shown in gray and oxygen atoms are shown in red.
Traditionally, S. divinorum has been ingested by chewing fresh leaves as a quid or by drinking an extract made from freshly crushed leaves. Alternatively, S. divinorum can be taken by pyrolizing dried leaves and rapidly inhaling the resulting smoke (3). The hallucinatory effects are potent and intense, with extraordinarily rapid onset, and can last up to an hour (3, 4). Interestingly, the hallucinations produced by S. divinorum appear to be qualitatively different from other hallucinogens; users typically describe their experience of “entering another reality” and having a “separation from body,” subjective descriptions that are consistent with a “spatiotemporal dislocation” (5–7).
Over the past decade, S. divinorum has become used for recreational purposes, particularly in the US and Europe. The use of S. divinorum as a legal hallucinogen has been facilitated by the availability of S. divinorum leaves and extracts through Internet suppliers. The use of S. divinorum has been unregulated, although several countries (e.g., Australia, Denmark, Italy, and Sweden) have recently classified S. divinorum as a controlled substance. In the US, S. divinorum is not listed under the Controlled Substances Act, although several states, including Missouri, Delaware, Louisiana, and Tennessee, have passed legislation controlling the use S. divinorum (see www.deadiversion.usdoj.gov).
The active component of S. divinorum is salvinorin A, a neoclerodane diterpene (3) (Figure 1⇑). The lipid-like salvinorin A molecule is chemically and structurally unique in that it represents the only known psychoactive diterpene and was the first non-nitrogenous hallucinogen to be identified. Smoking 200–500 μg of purified salvinorin A produces hallucinations that have been reported to be identical in nature to those observed following ingestion of fresh S. divinorum leaves (3). The effective dose of salvinorin A in humans is similar to that of the synthetic hallucinogens lysergic acid diethylamide (LSD) and 4-bromo-2,5-dime-thoxy-phenylisopropylamine (DOB) (8).
Initial attempts to identify the molecular target of salvinorin A were unsuccessful, despite binding probes against a number of molecular targets, including various serotonin (5-hydroxytryptamine; 5-HT) receptors (3). Subsequently, salvinorin A was submitted to the National Institutes of Mental Health Psychoactive Drug Screening Program (NIMH-PDSP; see http://pdsp.med.unc.edu) and screened against a panel of fifty molecular targets, including cloned human G protein–coupled receptors, transporters, and ion channels, and for comparison, the prototypical hallucinogen LSD was simultaneously screened (9). Surprisingly, salvinorin A displayed significant binding at the Gαi-coupled κ opioid receptor (KOR), but did not bind to the cloned δ opioid receptor (DOR) or μ opioid receptor (MOR) at the concentration tested. Salvinorin A also failed to bind to the human 5-HT2A receptor, the principal olecular target of classical hallucinogens such as LSD (9, 10). Functional studies measuring inhibition of forskolin-stimulated cyclic AMP accumulation and [35S]GTPγS binding confirmed salvinorin A as a potent agonist at cloned KOR and at the native KOR expressed in guinea pig brain (9). Salvinorin A thus became the first identified naturally occurring, non-nitrogenous KOR-selective agonist with psychotomimetic properties (9). This discovery has paved the road for a new avenue of opioid research and has fueled the search for other diterpenes with similar pharmacological properties.
Isolation of Salvinorin A and Related Diterpenes fromS. Divinorum
Although the psychotropic effects of Salvia divinorum have been widely known, the chemical component(s) responsible for these properties have not been extensively studied until quite recently. The first compounds isolated from S. divinorum leaves (Figure 2⇓) were the neoclerodane diterpene salvinorin A and salvinorin B (11) [structurally identical to the terpenoids independently isolated and designated elsewhere as divinorin A and divinorin B (1, 12)]. Salvinorin A in the leaves of S. divinorum ranges in concentration from 0.89 to 3.70 mg/g dry weight (13), and salvinorin B, as well as other isolated diterpenes, are found at much lower concentrations (14–16).
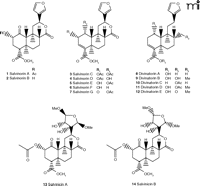
Structures of salvinorin A and B and related diterpenes isolated fromSaliva divinorum.
Salvinorin A was found to be a potent agonist at KOR, whereas salvinorin B, a potential metabolite of salvinorin A resulting from ester hydrolysis at the 2-acetoxy group, was inactive at KOR (9, 17). It has been suggested that rapid hydrolysis of salvinorin A resulting in the production of salvinorin B could contribute to the short duration of action in vivo (6). Chemical modifications at the C2 position have been implemented as a potential means of increasing the metabolic stability of salvinorin A (18) (Figure 3⇓). Intriguingly, salvinorin A is slightly more potent than other KOR-selective agonists, such as U50488H and U69593, in functional assays measuring potassium conductance through G protein–regulated K+ channels (17).
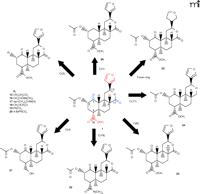
Structures of salvinorin A and representative derivatives. Representative modifications to the salvinorin A structure are shown for the indicated positions. Structural features of salvinorin A required for acitivty at KOR are shown in red, and elements shown in blue are not required for activity at KOR.
Following the identification of salvinorin A and B, several other diterpenes were isolated from the leaves of S. divinorum (Figure 2⇑): salvinorin C, which has weak affinity for KOR (19) and reportedly lacks psychotropic effects in humans (20); salvinorins D–G (15, 16, 21); divinatorins A–E (14, 21); and salvinicins A and B (22) (Figure 2⇑). The chemical structures and pharmacology of these salvinorin analogs have recently been characterized, and only salvinorin G (Ki = 418 ± 117 nM) and divinatorin D (Ki = 230 ± 21 nM) exhibit measurable affinities for KOR (19, 21, 23). Salvinorin A is the only isolated neoclerodane diterpene known to exhibit high affinity for KOR. Interestingly, initial studies indicate that salvinicin A is a partial agonist at KOR, whereas salvinicin B demonstrates weak antagonist activity at MOR (22). These observations suggest that modification of the unique scaffold of salvinorin A may lead to the discovery of novel selective agonists and antagonists.
Structure–Activity Relationships of Salvinorin A Derivatives
The functional groups of salvinorin A have been extensively modified, and subsequent structure activity relationship (SAR) studies have begun to clarify the pharmacophore. The rapidly expanding library of salvinorin A analogs includes modifications and substitutions at the C1 (19), C2 (17, 18, 24–28), C4 (18, 19), C17 (19), and C18 positions (18, 29), as well as the furan ring (19, 30) (Figure 3⇑). Subsequent SAR studies have been consistent with the pharmacophore proposed by Munro and colleagues (19, 23).
There are several reports exploring the effects of the 2-acetoxy group of salvinorin A on affinity and selectivity for KOR. In general, modifications in terms of size and electronegativity are not well tolerated at the C2 position (27). Interestingly, the propionate derivative retains high affinity for KOR (Figure 3⇑, structure 15) but exhibits partial agonist activity (17). Further SAR studies at the C2 position reveal that the N-methylacetamide and 2-epi-isopropylamine derivatives (Figure 3⇑, structures 16 and 17), which are predicted to have increased stability and aqueous solubility, are full agonists at KOR, with potencies comparable to salvinorin A (18). Additional work shows that substitution of the methoxymethyl group at the C2 position of salvinorin A (Figure 3⇑, structure 18) results in a full agonist with a sevenfold increase in potency at KOR (27). Derivatization of salvinorin A has also identified the C2 position as a critical site for receptor subtype selectivity. In fact, the 2-benzoate derivative (structure 19) was the first neoclerodane diterpene identified with MOR agonist activity (26). The 4-bromo benzoyl derivative (structure 20) also exhibits high affinity for MOR, whereas the affinity for KOR is decreased more than 350-fold, thus significantly increasing the selectivity for MOR over KOR (28). Intriguingly, the C2 epimer of salvinorin A (not shown) has decreased activity at KOR, but also exhibits weak antagonistic activity at DOR, identifying the first neoclerodane diterpene with DOR antagonist activity (29). These observations suggest that further modification of the salvinorin A scaffold may result in derivatives with improved receptor affinity and selectivity.
Modifications of salvinorin A at the C4 position reveal that the methyl ester group is required for KOR activity (Figure 3⇑, shown in red). The 18-hydroxy derivative (structure 21) fails to activate KOR but retains weak binding affinity (Ki = 347 ± 53 nM), perhaps representing antagonistic activity (19). Substitutions at the C18 position such as the dimethylamide derivative (structure 22), as well as other ester, amine, and ether substitutions, markedly decrease affinity for KOR (18, 29), confirming that the methyl ester group at the C4 position is a critical component of the pharmacophore. Removal of the lactone carbonyl from the C17 position (structure 24) has a nominal effect on the affinity and potency at KOR (19). Reduction of the C1 ketone to a hydroxy or acetoxy group significantly decreases binding affinity for KOR; however, removal of the ketone group (structure 26) causes only a fivefold decrease in binding affinity for KOR (19). The tetrahydro derivative (structure 25) displays a dramatic decrease (fortyfold) in affinity, but not potency, at KOR, suggesting that the furan ring is potentially part of the pharmacophore (19). Additional SAR studies reveal that epimerization at the C8 position (structure 23) results in a seventyfold decrease in binding affinity for KOR (19, 29). In summary, SAR studies suggest that the methyl ester at C4 and the furan ring at C12 are required for activity at KOR (Figure 3⇑, shown in red), whereas the C17 lactone and C1 ketone are not as stringently required (19) (Figure 3⇑, shown in blue).
Molecular Modeling and Mutagenesis Studies of the Salvinorin A:KOR Binding Complex
There have been a limited number of receptor mutagenesis and molecular modeling studies probing the salvinorin A binding pocket of KOR (9, 31, 32). Site-directed mutagenesis of KOR reveals that salvinorin A is stabilized in the binding pocket by interactions with tyrosine residues in helix 2 (i.e., Y119) and helix 7 (i.e., Y313 and Y320) (32). Molecular modeling predicts that Y119 in helix 2 and Y320 in helix 7 stabilize binding through hydrogen bonds with the furan ring of salvinorin A, and Y313 in helix 7 is predicted to stabilize binding through hydrophobic interactions with the 2-acetoxy group of salvinorin A (32) (Figure 4⇓). Mutation of Y139 in helix 3 and Y312 in helix 7 does not markedly alter the affinity of salvinorin A, suggesting that these residues are not involved in salvinorin A:KOR binding interactions (9, 32). In an elegant approach, Y119C, Y313C, and Y320C KOR mutants were exposed to a salvinorin A derivative containing a free sulfhydryl group (i.e., 2-thiosalvinorin B) (32). The affinity of 2-thiosalvinorin B was enhanced relative to salvinorin A at the Y313 mutant, suggesting that Y313 in helix 7 is in close proximity to the C2 position of salvinorin A (32). The key role of the conserved Y119, Y313, and Y320 residues in binding salvinorin A has been confirmed (31).

Proposed KOR:salvinorin A binding complex. Views are presented through the helical bundles (A) or from the extracellular side of KOR looking into the binding pocket of KOR (B). Salvinorin A is stabilized in the binding pocket through hydrogen bonding and hydrophobic interactions with Y119 (helix 2), Y313 and Y320 (helix 7), I294 (helix 6) (shown in green), and the second extracellular loop of KOR (shown in yellow). For clarity, some helical residues are not shown; figures were constructed using PyMOL.
In Vivo Pharmacology of Salvinorin: Implications for Human Therapeutics
Recently, there have been several reports verifying that salvinorin A exerts its effects via KOR activation in vivo. Thus, intraperitoneal injections of salvinorin A (1.0 – 4.0 mg/kg) results in a dose- and time-dependent increase in tail-flick latencies in mice, with the maximal response occurring within ten minutes of salvinorin A administration (33, 34). Thereafter, the antinociceptive effects of salvinorin A diminish quite rapidly and return to baseline within thirty minutes of drug administration (34), which may reflect rapid metabolism of salvinorin A in vivo (6, 48). These antinociceptive effects were also observed using the hotplate assay, in which increased latencies occur at a salvinorin A dose of 1.0 mg/kg (34). Salvinorin A effects on tail-flick latencies are abolished following pretreatment with the KOR-selective antagonist nor-bin-altorphimine, demonstrating the selectivity of salvinorin A for the KOR in vivo (33, 34). Salvinorin A also exhibits dose- and time-dependent antinociceptive effects in the chemonociceptive acetic acid abdominal constriction assay (34).
The antinociceptive effects of salvinorin A and the 2-propionate derivative (Figure 2⇑, structure 15) have also been examined in wild-type and a novel strain of KOR knockout mice (34). In these studies, salvinorin A and 2-salvinorinyl propionate produced concentration- and time-dependent antinociception in wild-type mice but not in the KOR knockout mice. Similar to other KOR-selective agonists (36), salvinorin A and the 2-propionate derivative reduce rectal body temperature in wild-type mice, but this effect is absent in KOR knockout mice (35). Salvinorin B tested at the same concentrations failed to produce antinociception or hypothermic effects in wild-type mice, which is consistent with in vitro observations demonstrating that salvinorin B is inactive at KOR (9, 17).
In addition to the recognized potential for antinociceptive therapy, it has recently been suggested that S. divinorum may provide a pharmacological basis for the treatment of diarrhea (37). Specifically, salvinorin A, as well as extract prepared from S. divinorum leaves, can inhibit myenteric cholinergic transmission in guinea pig ileum. The inhibitory effects of salvinorin A on ileum contraction are blocked by the non-selective opioid receptor antagonist naloxone and the KOR-selective antagonist nor-BNI. Taken together, these behavioral observations suggest that salvinorin A exerts its effects via activation of KOR in vivo.
There is considerable evidence supporting a role for the KOR:dynorphinergic signaling complex in the regulation of mood disorders (9, 38, 39). Indeed, KOR agonists induce depressive-like behaviors in animal models, whereas KOR antagonists have antidepressant-like effects in vivo (40). These observations have led, in part, to the hypothesis that modulation of KOR signaling pathways will be useful for the treatment of depressive behaviors (41) (Figure 5⇓). There is also significant evidence to support the involvement of KOR signaling pathways in the dependence of cocaine [for review, see (49)].To this end, several studies have examined whether salvinorin A can elicit the same behavioral responses as other synthetic KOR-selective ligands.
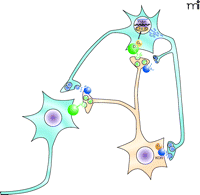
Potential role of the KOR–dynorphinergic signaling complex in the treatment of mood disorders. This figure depicts the mesolimbic reward pathway that has been implicated in mood disorders. Dopaminergic neurons project from the ventral tegmental area (orange) to GABAergic neurons in the nucleus accumbens (blue). Synaptic dopamine (green triangles) release can lead to activation of Gαs-coupled D1 dopamine receptors (green spheres) resulting in accumulation of cyclic AMP, and subsequent activation of the cAMP response element binding protein (CREB) that regulates transcription of dynorphin (blue rectangles), the endogenous KOR ligand (46). In this model, dynorphin activates the presynaptic KOR that modulates dopamine release in the nucleus accumbens. Stress-induced increases in CREB activity have been associated with depressive-like behaviors in animal models, similar to the behavioral effects observed with administration of KOR-selective agonists such as salvinorin A, which decrease extracellular concentrations of dopamine within the nucleus accombens. These CREB-induced depressive behaviors can be attenuated with KOR-selective antagonists (47), further implicating a role for the KOR-dynorphinergic signaling pathway in the treatment of depressive-like behaviors.
Intraperitoneal injections of salvinorin A (0.25 – 2.0 mg/kg) in rats result in a dose-dependent increase in immobility in the forced swim test without altering locomotor activity in an open field (41). The same concentration range increases the threshold for intracranial self-stimulation and decreases extracellular dopamine concentrations in the nucleus accumbens (41). These observations are consistent with effects of the KOR-selective agonist U69593 (40) and support the hypothesis that the KOR system may play a critical role in depressive-like behaviors. It is noteworthy, however, that there has been at least one report demonstrating the usefulness of S. divinorum in the treatment of refractory depression (42). Ultimately, clinical trials will be needed to resolve the potential role for selective KOR agents in treating mood disorders.
Additional studies in mice reveal that salvinorin A decreases extracellular dopamine levels in the caudate putamen, but not in the nucleus accumbens (43). The effect can be abolished by pretreatment with the KOR-selective antagonist nor-BNI (43). Salvinorin A also leads to conditioned place aversion and a decrease in locomotor activity (43), which is consistent with the observation that salvinorin A disrupts climbing behavior in an inverted screen task (44). These studies suggest that salvinorin A causes some level of sedation and motor incoordination.
Butelman et al. also have demonstrated that salvinorin A exerts its effects via activation of KOR in vivo. In non-human primates, salvinorin A produces a discriminative stimulus similar to the KOR-selective agonist U69593, whereas the NMDA antagonist ketamine is not generalized by U69593-trained subjects; the effect of salvinorin A can be blocked by a KOR-selective antagonist (45). Taken together, these animal studies are consistent with in vitro pharmacology studies demonstrating that salvinorin A is a highly potent and selective KOR agonist, which likely accounts for the effects of the drug in humans.
Conclusions
Salvinorin A represents the first known non-nitrogenous KOR selective agonist and the first non-alkaloidal hallucinogen. Salvinorin A is a potent KOR agonist in vitro and in vivo, suggesting that the hallucinogenic effects produced by salvinorin A are mediated by activation of KOR. Modification of the salvinorin A scaffold has also led to the development of MOR-selective agonists. Thus, the chemical structure of salvinorin A may be manipulated for the design of novel receptor-specific ligands. Further studies with kappa agonists and antagonists will likely provide insight into the therapeutic potential of the KOR–dynorphinergic system in the treatment of psychiatric disorders associated with hallucinations, such as schizophrenia and Alzheimer’s disease, as well as various mood disorders.
Acknowledgments
This work was supported by NIDA grants RO1 DA017204 (BLR) and NO1 MH80004 for the NIMH-PDSP (BLR). Timothy Vortherms has been awarded a postdoctoral National Research Service Award from NIDA (1F32 DA022188). The authors would like to thank Dr. Richard Westkaemper for providing the salvinorin A:KOR binding model and Daniel Siebert for the Saliva divinorum photograph. The authors would also like to thank Ryan Strachan for careful review and helpful comments with this paper.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Timothy A. Vortherms, PhD, received his doctoral degree from the Department of Medicinal Chemistry and Molecular Pharmacology at Purdue University in 2004. He is currently a postdoctoral fellow and has received a Ruth L. Kirschstein National Research Service Award to study the molecular determinants for salvinorin A interactions with κ opioid receptors.

Bryan L. Roth, MD, PhD, is a Professor in the Department of Pharmacology, School of Medicine and Division of Medicinal Chemistry and Natural Products, School of Pharmacy at the University of North Carolina at Chapel Hill. He is also Director of the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH-PDSP). His research interests include GPCR structure and function, and he is actively involved in drug discovery using the resources of the NIMH-PDSP. Address correspondence to BLR. E-mail bryan_ roth{at}med.unc.edu; fax 919-843-5788.

