Collateral Damage in Cancer Chemotherapy: Oxidative Stress in Nontargeted Tissues
Abstract
Injury to nontargeted tissues in chemotherapy often complicates cancer treatment by limiting therapeutic dosages of anticancer drugs and by impairing the quality of life of patients during and after treatment. Oxidative stress, directly or indirectly caused by chemotherapeutics as exemplified by doxorubicin, is one of the underlying mechanisms of the toxicity of anticancer drugs in noncancerous tissues, including the heart and brain. A comprehensive understanding of the mechanisms of oxidative injury to normal tissue will be essential for the improvement of strategies to prevent or attenuate the toxicity of chemotherapeutic agents without compromising their chemotherapeutic value.

Introduction
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are generated in vivo from dioxygen (O2) and nitric oxide (NO), respectively. It is somewhat paradoxical that the reactive natures of both gases, which lies at the core of essential life processes, also engenders derivatives (i.e., certain ROS and RNS) that can be deleterious to life; such derivatives include superoxide radical (O2·−), hydrogen peroxide (H2O2), hydroxyl radical (HO·), alkoxyl/peroxyl radicals (RO·/ROO·), and peroxynitrite (ONOO−). The medical relevance of ROS and RNS is made further complex in that a great diversity of biophysical processes and conditions can support their production. For example, O2·− is primarily produced by incomplete reduction of O2 (1), whereas NO is generated from l-arginine via a nitric oxide synthase–catalyzed reaction (2). Many ROS/RNS are crucial signaling molecules, tightly regulated and essential to crosstalk mechanisms among multiple cellular pathways (3). In order to check the activities of ROS/RNS in vivo and maintain cellular redox conditions, antioxidant systems have evolved that consist of biological antioxidants (e.g., vitamin C, vitamin E, and glutathione) and antioxidant enzymes (e.g., super-oxide dismutase, catalase, and glutathione peroxidase). When the generation of ROS/RNS exceeds cellular adaptive and repair capacities—a condition that is referred to as oxidative stress—biological molecules such as nucleic acids, proteins, and membrane phospholipids become damaged through oxidative reactions. Oxidative stress results in the failure of normal cellular functions and even cell death.
The US Food and Drug Administration (FDA) has approved 132 anticancer drugs (a full list is available at http://www.fda.gov/cder/cancer/approved.htm). Fifty-six of these have been reported to induce oxidative stress, including the anthracyclines, cyclophosphamide, cisplatin, busulfan, mitomycin, fluorouracil, cytarabine, and bleomycin (Figure 1⇓). Some chemotherapeutic agents, such as bleomycin (4), induce oxidative stress as a mechanism for killing cancer cells. However, certain chemotherapeutic agents, such as the anthracyclines (5–7), induce oxidative stress in nontargeted tissues and thereby lead to “normal tissue injury.” Although chemotherapy improves the survival rates of cancer patients, oxidative stress–mediated impairment of normal tissues is a significant side effect and decreases the quality of life of patients. A better understanding of the mechanisms involved in oxidative injury to normal tissues is essential to the design of intervention strategies that will attenuate the toxicity of chemotherapeutic agents without compromising their anticancer efficacy.
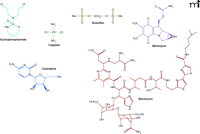
Anticancer drugs that cause oxidative stress. Cyclophosphamide and cisplatin are converted to reactive metabolites that cause DNA cross-linking and react with the thiol groups in enzymes and glutathione. Busulfan decreases glutathione levels. Mitomycin induces ROS generation through redox cycling of the quinone moiety. Cytarabine incorporates into DNA and acts as a chain terminator; following cytarabine-induced damage to DNA, ROS are generated in vivo. Bleomycin forms complexes with Fe2+ and O2 that break DNA strands and generate superoxide and hydroxyl radicals. In all, 56 of 132 FDA-approved anticancer drugs have been implicated in oxidative stress (as determined through Scinfinder Scholar using each drug name and the term “oxidative stress” as keywords). See text for details.
To address the problem of oxidative damage in normal tissues exposed to chemotherapeutics, we have studied doxorubicin (DOX, also called adriamycin), a representative of the anthracyclines, which remain one of the most effective anticancer drug families in clinical use. Other anthracyclines used in clinical practice include daunorubicin, epirubicin, idarubicin, aclarubicin, pirarubicin, and mitoxantrone (Figure 2⇓). DOX kills cancer cells through DNA intercalation and inhibition of topoisomerase II (8), but its side effects include oxidative stress–mediated injuries to heart (5), kidney (6), and brain (7). DOX nevertheless remains an important component in most chemotherapeutic regimens, owing to its efficacy in treating a broad spectrum of cancers.
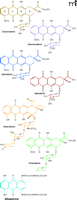
Anthracyclines in current clinical use. Doxorubicin and daunorubicin are two prime anthracyclines used as anticancer drugs. Doxorubicin is used in treatment of breast cancer, aggressive lymphomas, childhood solid tumors, and soft tissue sarcomas, whereas daunorubicin is used to treat acute lymphoblastic or myeloblastic leukemias. Structural analogs of doxorubicin and daunorubicin (i.e., epirubicin, pirarubicin, idarubicin, aclarubicin, and mitoxantrone) were developed in the search for chemotherapeutics with limited cardiotoxicity; the success of this search has been rather modest. See text for details.
Although many tissues are susceptible to the unintended effects of chemotherapy, here we will focus on heart and brain. Because cardiomyocytes and neurons are post-mitotic cells, major insults that befall them are generally irreversible and thus can irrevocably affect the functions of the heart and brain. Cardiotoxicity is well recognized in chemotherapy (9), especially in patients treated with anthracyclines, which can cause the dose-dependent development of dilated cardiomyopathy and congestive heart failure (10). Recent studies have revealed that brain tissue is similarly susceptible to chemotherapeutic agents, despite suppositions of protection via the blood–brain barrier (BBB) (11). Persistent changes in cognitive function, including memory loss, distractibility, and difficulty in performing multiple tasks, have been observed in breast cancer survivors after treatment with chemotherapeutic agents, including DOX (11–13). The term “chemobrain” has been used to describe the cognitive decline associated with chemotherapy (14, 15). Moreover, magnetic resonance imaging has revealed that chemotherapy has an influence on the structures of the brain (16–18), which might be related to cognitive impairments. In addition, positron emission tomography has shown defects in brain function in breast cancer patients five to ten years after chemotherapy (19, 20). Although the molecular basis for “chemobrain” remains elusive, oxidative stress has been thought to play a mechanistic role (7, 11, 21–23).
ROS/RNS Generation and Disturbance of Redox Status in Chemotherapy
Oxidative stress results from an imbalance between the generation of ROS/RNS and their removal by the cellular antioxidant system. The heart and brain manifest distinct mechanisms of DOX-induced ROS/RNS production. In heart, DOX-induced production of ROS/RNS involves the interplay of multiple processes, including redox cycling of the quinone moiety of DOX, disturbance of iron metabolism, and DOX metabolites. In contrast, tumor necrosis factor-α (TNF-α) has been implicated in the generation of ROS/RNS in the brain (21).
Mechanisms of Toxicity in the Heart
The heart is rich in mitochondria, which occupy about forty percent of the total intracellular volume of cardiomyocytes (24). DOX has a high affinity for cardiolipin, a negatively charged phospholipid abundant in the mitochondrial inner membrane (25), leading to mitochondrial accumulation of DOX (26). Under clinically relevant plasma DOX concentrations of 0.5–1 μM, intramitochondrial concentrations can reach approximately 50–100 μM. At these relatively high concentrations of DOX, the heart becomes a site of redox reactivity. Specifically, the quinone functionality of DOX is transformed, in the presence of NADH, into a semiquinone via one-electron reduction by complex I of the electron transport chain (27). The semiquinone form reacts with molecular oxygen to produce a superoxide radical (O2·−), whereby DOX returns to the quinone form. The cycling of DOX between qui-none and semiquinone forms generates large amounts of O2·− (Figure 3⇓), which further give rise to a variety of active ROS/RNS species, including H2O2, ·OH, and ONOO− (28, 29). In heart tissue, several endogenous reductases (30) and the endothelial isoform of nitric oxide synthase (31) can catalyze the redox cycling of DOX.
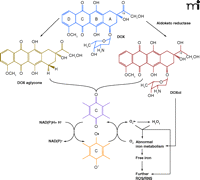
DOX-induced generation of ROS/RNS in heart. Cleavage of the sugar residue and reduction of the carbonyl group at C13 produce DOX aglycone and doxorubicinol (DOXol), respectively. Redox cycling of DOX and its metabolites generates ROS including O2·− and H2O2. ROS and DOXol can affect iron metabolism and lead to increased levels of cellular free iron ions, which induce further ROS/RNS generation.
DOX also interferes with normal metabolic reactions that involve iron, a redox-active transition metal, again leading to ROS generation (Figure 3⇑). DOX semiquinone, O2·−, and their byproduct H2O2 can trigger the release of iron from ferritin, an important iron storage protein (10), as well as from cytoplasmic aconitase, which contains a [4Fe-4S] cluster. Loss of the [4Fe-4S] cluster converts cytoplasmic aconitase into iron regulatory protein (IRP)-1. IRP1 binds with high affinity to conserved iron-responsive elements (IRE) in the untranslated regions of transferrin receptor (TfR) and ferritin mRNAs, stabilizing TfR mRNA but destabilizing ferritin mRNA (32, 33). As a consequence, iron uptake exceeds iron sequestration, so that increased cellular levels of free iron lead to production of ·OH via Fenton chemistry, contributing to oxidative stress and cytotoxicity.
Metabolic turnover of DOX per se is another factor leading to ROS generation. Aldoketo reductases convert the carbonyl group at C13 to a hydroxyl group, thereby conferring a secondary alcohol function upon DOX (Figure 3⇑) (10). The secondary alcohol derivative of DOX is effective in releasing iron from the [4Fe-4S] cluster of cytoplasmic aconitase (34, 35), resulting in further disturbance of iron metabolism and further oxidative stress (36). DOX can also be metabolized into its aglycone form, which results from cleavage of the sugar residue off the parent compound (37). The lipophilic aglycone metabolite of DOX has a higher membrane diffusion capacity than its parent compound and can accumulate in the mitochondrial inner membrane. Diversion of electrons from the respiration chain leads to ROS formation and deterioration of the functional integrity of the respiration chain.
Mechanisms of Toxicity in the Brain
In comparison with the multiple mechanisms proposed for DOX-induced ROS/RNS generation in the heart, much less is known about such mechanisms in the brain (11, 21, 22). It is generally believed that ATP-dependent transporters at the BBB prevent passage of DOX through the barrier (38), and indeed DOX has not been detected in the areas protected by the BBB, such as the cortex and the hippocampus (21, 39). In contrast, we have found increased levels of TNF-α in the cortex and the hippocampus of mice treated with DOX (21). Moreover, markers of oxidative stress, including protein carbonyl levels, protein-conjugated 4-hydroxy-2-nonenal (4-HNE; a product of lipid peroxidation), and protein nitrotyrosine are elevated in the brains of DOX-treated mice (22). It is therefore likely that oxidative stress in the brains of these animals results from indirect effects of DOX and that TNF-α is a mediator of DOX-induced ROS/RNS. Administration of DOX has also been shown to increase circulating levels of TNF-α (7, 21, 40). Circulating TNF-α can pass the BBB (41) and activate glial cells to initiate local production of TNF-α (42), which in turn induces nitric oxide synthase, leading to the generation of RNS (Figure 4⇓) (7). Furthermore, the role of nitric oxide synthase in RNS production in the brain during DOX treatment is strongly supported by two independent investigations in rat models. In one study, daunorubicin, an analog of DOX (Figure 2⇑), raises the level of nitric oxide synthase in the brain (43), whereas inhibition of nitric oxide synthase by aminoguanidine ameliorates DOX-induced oxidative stress in the brain (44). The role of circulating TNF-α in mediating ROS/RNS is supported by the fact that co-administration of anti-TNF-α antibody with DOX completely prevents TNF-α elevation and DOX-induced brain injury (21).
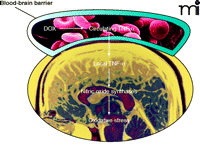
Proposed mechanism of DOX-induced oxidative stress in brain. DOX leads to increased levels of circulating TNF-α, which can pass the blood-brain barrier and trigger local production of TNF-α in the brain. Increased levels of TNF-α may in turn induce the expression of nitric oxide synthases, leading to oxidative stress.
Cellular Modulation of Oxidative Stress
Cellular antioxidants and antioxidant enzymes play a vital role in scavenging ROS/RNS and maintaining a balanced cellular redox status. ROS/RNS that arise during chemotherapy consume cellular antioxidants and lead to oxidative modification and inactivation of antioxidant enzymes. This cascade of events pushes the cellular redox status towards the direction of oxidative stress.
Glutathione, a ubiquitous thiol-containing tripeptide, functions directly as an antioxidant in vivo. Glutathione-dependent enzymes, such as glutathione peroxidase, glutathione reductase, and glutathione-S-transferase, utilize glutathione to neutralize ROS (45). Glutathione acts as a reductant to convert hydrogen peroxide into water and to reduce lipid peroxides to their corresponding alcohols; in these reactions, two molecules of glutathione must combine to form a disulfide bond (i.e., GSSG), which is catalyzed by glutathione peroxidase. To complete this redox cycle, glutathione reductase uses NADPH to catalyze the conversion of GSSG back into two molecules of glutathione. In addition, glutathione, by virtue of its nucleophilic thiol group, can conjugate directly with lipid oxidation products or xenobiotics to produce thioethers; these adduct reactions are catalyzed by glutathione-S-transferase and provide important mechanisms for the detoxification and excretion of toxic substances. The ROS that are produced in response to chemotherapeutic agents such as DOX decrease cellular glutathione reserves (44) and thereby escalate oxidative stress (46, 47). DOX administration further leads to a dose-dependent decrease of glutathione peroxidase activity in the heart (48) and glutathione-S-transferase activity in the brain (23); decreases in these enzyme activities presumably reflect mechanisms of oxidative stress. In brain tissue of DOX-treated mice, manganese superoxide dismutase (MnSOD), an essential mitochondrial antioxidant enzyme, has been found to be inhibited upon nitration (7). Tyr34 is vital for MnSOD activity, and its nitration impedes substrate binding (49).
ATP-dependent transporter proteins such as multidrug resistance–associated proteins (MRPs) also play important roles in maintaining the cellular redox balance by transporting glutathione-conjugated lipid oxidation products out of cells (50). We found, in the heart tissue of DOX-treated mice, that 4-HNE modifies and inhibits Mrp-1, thereby exacerbating DOX-induced oxidative stress (51). Moreover, levels of Mrp-1 are elevated in the brains of DOX-treated mice (22). Given that MRPs, together with P-glycoprotein (i.e., MDR1), are intrinsically expressed at the BBB to regulate drug distribution and efflux (52), these findings prompt the intriguing question as to whether chemotherapy-based inactivation of ATP-dependent transporter proteins compromises the integrity of the BBB, leading to drug accumulation in the brain and thus contributing to further oxidative stress.
Routes to Oxidative Damage of Nontargeted Tissues
DOX affects proteins at both the transcriptional and post-translational levels. Microarray analysis has shown that DOX can modulate the expression of cardiac proteins related to energy production and metabolism, calcium regulation, cell growth and death, cytoskeletal function, cell adhesion, and signal transduction (53). Among the affected proteins, enzymes involved in bioenergetic and metabolic pathways are of particular importance, because the maintenance and functionality of the heart require great inputs of energy (53–55). Disruption of metabolic pathways that normally support energy production, energy transfer, and energy utilization directly contribute to heart injury. At the transcriptional level, DOX decreases the expression of phospholipase Cδ , a crucial enzyme for phosphoinositol signaling and calcium homeostasis (53). At the post-translational level, DOX-induced ROS can oxidize a susceptible cysteine residue (i.e., Cys278) of mitochondrial creatine kinase, thereby inactivating this pivotal, energy-regulating enzyme (54). Oxidative modification has also been implicated in the inhibition of NADH dehydrogenase in the heart (27). Through an emerging redox proteomic approach that combines the power of two-dimensional electrophoresis and immunochemistry, proteins with elevated oxidative modification markers, such as carbonyl levels, 4-HNE, and nitrotyrosine, have been identified (56). In a mouse model, redox proteomics showed that cardiac triose phosphate isomerase, β-enolase, and electron transfer flavoprotein-ubiquinone oxidoreductase (ETF-QO) become carbonylated after acute DOX treatment (55). Triose phosphate isomerase and β-enolase operate within the glycolytic pathway: the isomerase catalyzes the interconversion between glyceraldehyde-3-phosphate and dihydroxyacetone phosphate, and β-enolase catalyzes the conversion of 2-phosphoglycerate to phosphoenolpyruvate. ETF-QO is an iron–sulfur ([4Fe-4S]) flavoprotein, located at the inner mitochondrial membrane, that catalyzes electron transfer from ETF to co-enzyme Q, a process crucial to the metabolic oxidation of certain fatty acids and amino acids. Consequently, the proteomic identification of cardiac proteins that are inactivated following DOX treatment (55) suggests that ATP production would be compromised in heart, a suggestion that is indeed consistent with observed effects of DOX.
At the subcellular level, mitochondria are the main targets of DOX-induced oxidative stress. Electron micrographs clearly demonstrate that DOX causes profound damage to the organelle, including mitochondrial vacuolization, mitochondrial degeneration, and disruption of the mitochondrial membrane. Mitochondrial DNA (mtDNA) is also an important target for oxidative stress. The circular molecule of mtDNA within the human organelle consists of 16,569 base pairs (57) and encodes two rRNAs, twenty-two tRNAs, and thirteen polypeptides, all of which are essential to oxidative phosphorylation (58). Compared with nuclear DNA (nDNA), mtDNA is more susceptible to oxidative damage, owing to its proximity to the site of ROS generation, its lack of introns and histones, and the limited DNA repair capacities in mitochondria (59). Consequently, the mutation rate of mammalian mtDNA is ten- to seventeenfold higher than that of nDNA (60). In the heart, DOX-induced oxidative stress causes formation of 8-hydroxydeoxyguanosine (61) and mtDNA deletions (62, 63); significantly, levels of 8-hydroxydeoxyguanosine persist after termination of DOX treatment (64). The heightened, oxidation-sensitive mutability of mtDNA has obvious implications for cellular energy production. Upon accumulation of insults to mtDNA, cellular capacities for energy production cannot meet the constant physical demands placed on the heart, and cardiac hypertrophy and heart failure ensue (65). In the brain, there is no direct evidence of mtDNA damage in DOX treatment, but research into this question has only just begun (7, 21). Intriguingly, age-related neurodegenerative diseases such as Parkinson disease and amyotrophic lateral sclerosis are characterized by mtDNA oxidative damage and neuronal dysfunction (66). It is tempting to speculate that mtDNA oxidative damage might play a vital role in the cytotoxicity of DOX in the brain.
Protection against Chemotherapy-Induced Oxidative Damage
Total cumulative dose, rate of administration, duration of chemotherapy, and concomitant use of other cardiotoxic drugs are well-recognized risk factors for DOX cardiotoxicity (67). Several strategies have been applied to mitigate the side effects of DOX. The maximum cumulative dose of DOX is 450–550 mg/m2 (68); there is some evidence that slow (i.e., over 48 or 96 hours) continuous infusion of DOX may be less cardiotoxic in adults (69) [although not in children (70)]. Otherwise, intensive research has shown only moderate success in finding analogs of DOX that have equivalent anticancer activity but less toxicity. Among DOX analogs, epirubicin has a higher cumulative dose than DOX, but its activity against breast cancer is lower (71, 72); idarubicin is a more potent anticancer drug, but its cardiotoxicity is no less than DOX (73, 74); mitoxantrone also has cardiotoxicity similar to DOX. These analogs still manifest toxicity in brain tissue (75). (See Figure 2⇑ for structures.)
Insights into the key steps mediating DOX-induced oxidative stress have suggested mechanism-based strategies to prevent normal tissue damage (Figure 5⇓). One strategy is to disrupt the ROS propagation chain. Dexrazoxane (DRZ), an iron chelator, has been cooperatively used with DOX to chelate the free iron ions released by DOX and its metabolites, and thus to inhibit Fe2+-related ROS generation without interfering with the anticancer potency of DOX (76). DRZ has demonstrated efficacy in ameliorating both acute and chronic DOX cardiotoxicity in mammalian models including mouse, dog, swine (77), and in human cancer patients (76). A recent report shows, however, that DRZ may be risky to use with certain cancers (78), demonstrating the necessity for novel intervention methods. The discovery that TNF-α mediates the toxicity of DOX in the brain suggests that an anti-TNF-α antibody could be a potential modality to quench oxidative stress in the brain, as recently demonstrated by the preliminary success of the proof-of-concept study in a mouse model (21). In addition, the insight that isoforms of nitric oxide synthase may be involved in RNS production and oxidative stress in the brain (7, 79, 80) suggests that selective inhibition of these enzyme activities could be therapeutic (44).
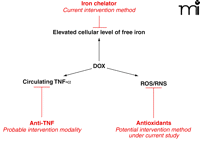
Mechanism-based interventions into DOX-induced oxidative damage. Iron chelators reduce free cellular iron ions and disrupt the propagation of ROS/RNS and are thus used to reduce DOX cardiotoxicity. The efficacies of antioxidants to decrease oxidative damage in the heart and brain are now also under intensive study. Modulation of circulating TNF-α may be an intervention modality to reduce DOX-induced oxidative damage in the brain. See text for details.
The efficacy of antioxidant enzymes to reduce DOX-induced cardiac injuries has been demonstrated in transgenic mice that overexpress MnSOD, catalase, metallothionein, or thioredoxin-1 (81–84). These valuable transgenic animal models clearly warrant further characterization and may reveal possible mechanisms by which these antioxidant enzymes protect the brain from DOX toxicity. Intervention strategies may include direct use of antioxidant enzyme mimetics. A cell-permeable SOD mimetic (i.e., MnTBAP) and a glutathione peroxidase mimetic (i.e., Ebselen) have been shown to protect cardiomyocytes from DOX-induced toxicity (85).
Co-administration of antioxidants or antioxidant precursors is another way to diminish oxidative stress by direct removal of DOX-induced ROS. Recently, grape seed proanthocyaniclins, a dietary antioxidant supplement, have been shown to enhance the anti-tumor activity of DOX and ameliorate DOX-induced myocardial oxidative stress in tumor-bearing mice (86). Pretreatment of mice with gamma-glutamyl-cysteine ethyl ester, a precursor of glutathione, increases brain glutathione levels and significantly decreases glutathione-S-transferase–facilitated protein oxidation and lipid peroxidation (23). These results demonstrate the feasibility of co-administration of antioxidants as a strategy to prevent nontargeted tissues from DOX-induced oxidative stress.
Summary
Oxidative stress is one mechanism by which many chemotherapeutic agents kill cancer cells. As a side effect, however, these chemotherapeutic agents can also place nontargeted (i.e., noncancerous) tissues under conditions of oxidative stress and thereby undermine the health of organs such as the heart and brain. Such damage not only limits the effective dosage of chemotherapeutics, but also compromises the quality of life of cancer patients after chemotherapy. Chemotherapy-induced ROS/RNS species [and their concomitant oxidative damage to proteins (including anti-oxidant and energy-generating enzymes), lipids, nucleic acids, and larger cellular components (e.g., membranes and mitochondria)] are prime suspects in the toxic side effects of acute or chronic chemotherapeutic treatment. Intensive investigations to pursue the relationship between oxidative stress–generating anticancer drugs and damage to nontargeted tissues are greatly benefiting from modern technologies, including proteomic techniques. Novel intervention strategies to mitigate the potential side effect of chemotherapeutics on normal tissues are increasingly relying on our mechanistic insights into the roles of chemotherapeutics in promoting cellular oxidative stress.
Acknowledgments
This work is supported by NIH grants CA 49797 and CA 94853.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Yumin Chen, PhD, is a postdoctoral scholar in the Department of Toxicology at the University of Kentucky. He performed his doctoral work in Biochemistry at Miami University. His research interests pertain to the biochemistry and toxicity of ROS/RNS. Paiboon Jungsuwadee, PhD, is a senior scientist in the Department. He received his degree in Pharmacology and Toxicology from the University of Vienna, Austria. His research interests span pharmacology and immunology. Daret St. Clair, PhD, is the James Graham Brown Foundation Endowed Chair in Neuroscience and Professor of Toxicology at the University of Kentucky. She has been active in the field of free radicals in biology and medicine for over fifteen years. Her research interests pertain to antioxidant enzymes, especially superoxide dismutase and its role in cancer. Mary Vore, PhD, is Professor and Chair of the Department of Toxicology at the University of Kentucky. Her studies address mechanisms of drug metabolism and detoxification, with a focus on hepatic transporters, their role in the elimination of xenobiotics, and the metabolism of endogenous substrates such as steroid conjugates and bile acids. Allan Butterfield, PhD, is the Alumni Professor of Biological Chemistry in the Department of Chemistry at the University of Kentucky. His expertise is in the field of oxidative stress and brain injury. His research has been focused on protein oxidation and its role in the development of diseases associated with neurodegeneration. Address correspondence to DSC. E-mail dstcl00{at}uky.edu; fax 859-323-1059.

