Microsomal Prostaglandin E2 Synthase: A Safer Target than Cyclooxygenases?
For over a century, aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) have been extensively used in the treatment of prostaglandin (PG) E2-mediated inflammatory diseases [e.g., rheumatoid arthritis (RA)] and pain-related disorders. Traditional (t)NSAIDs influence PG-mediated pathological and physiological processes by non-selectively inhibiting the activities of PG G/H synthase (PGHS)-1 and -2, also referred to as cyclooxygenase (COX)-1 and -2, respectively. These enzymes catalyze the rate-limiting step of prostaglandin synthesis from arachidonic acid (Figure 1⇓). The most commonly prescribed tNSAIDs are naproxen, ibuprofen, and diclofenac. Although these tNSAIDs inhibit both COX isoforms, their selectivity for COX-2 varies: naproxen has relatively low selectivity for COX-2, and diclofenac relatively high. According to the classical paradigm, COX-1 is constitutively expressed and mediates the and other PGs which regulate organ homeostasis formation of PGE2 and physiological “housekeeping” functions, such as gastric cytoprotection and hemostasis. The expression of COX-2, on the other hand, is inducible and increases in response to proinflammatory cytokines. The discovery of constitutive COX-2 expression in brain and renal tissue and the finding that COX-1-derived prostaglandins can also contribute to inflammation, however, changed this somewhat simplistic subdivision of the COX isoforms. Nonetheless, this classical hypothesis served as the basis for the development of selective COX-2 inhibitors (coxibs) in the early nineties, which have comparable anti-inflammatory properties to tNSAIDs, but have the advantage over the older drugs that the production of “housekeeping” PGs remains intact. These new drugs provide a means by which the serious gastrointestinal side effects associated with the use of tNSAIDs could be bypassed. Indeed, rofecoxib (Vioxx) reduced the incidence of serious adverse gastrointestinal events from 4% (as observed with administered naproxen) to 2% in patients with RA (1). Celecoxib, with less selectivity for COX-2 as compared to rofecoxib, also exhibited fewer gastrointestinal side effects as compared to those observed with the older tNSAIDs diclofenac and ibuprofen after six months (2), although longer follow up data revealed no significant difference (3). These results, together with effective marketing directly to consumers, resulted in a rapid increase of prescriptions worldwide of coxibs, which soon dominated the market for NSAIDs. Several years later, however, it became evident that rofecoxib increased the risk of major adverse cardiovascular events, such as myocardial infarction and stroke, in a trial designed to compare rofecoxib to placebo for its potential to reduce the incidence of colorectal adenomas (4). Interestingly, a higher incidence of cardiovascular events had been found earlier, but the difference was thought to arise from possible protective effects of naproxen rather than harmfulness of rofecoxib. The use of celecoxib also became associated with increased cardiovascular risk (5), suggesting a class-wide effect, although meta-analysis of the available trials revealed that celecoxib in commonly used doses might not increase vascular risk (6). These discoveries prompted active discussion as to whether the beneficial effects (i.e., gastroprotection, endothelial function, and atherosclerosis) outweighed the known harmful effects [i.e., thrombosis, hypertension, and adverse cardiac remodeling (7)] of COX-2 inhibitors and whether the use of coxibs should be continued in patients at risk for cardiovascular disease. Indeed, the controversy even led to withdrawal from the market of rofecoxib, by that time the most frequently prescribed COX-2 inhibitor. Recent studies also raise serious questions about the cardiovascular safety of diclofenac, a tNSAID with relatively high COX-2 selectivity as compared to that of naproxen or ibuprofen (8, 9). An evidence-based debate is ongoing that should facilitate the difficult decision-making process of effective anti-inflammatory therapy with NSAIDs while minimizing cardiovascular and gastrointestinal risk. Concomitant use of low-dose aspirin seems to decrease cardiovascular risk of coxibs (10), but the combined use of aspirin with any NSAID increases the risk of gastrointestinal complications. A non-selective NSAID with low cardiovascular risk, such as naproxen, in combination with a proton-pump inhibitor (PPI), may be the current preferred choice of treatment. Additional information, however, from adequately designed clinical trials comparing COX-2 inhibitors and tNSAIDs––both with and without aspirin and PPIs––in patients with limited risk and in patients with increased cardiovascular or gastrointestinal risk, will be essential for making well-considered clinical decisions in the future.
The biological basis for increased cardiovascular risk associated with COX-2 inhibitors had been thought to arise from a disruption of the balance between platelet and endothelium COX-2- COX-1-derived thromboxane (Tx)A2 is a vasoconstrictor and a derived prostacyclin (PGI2) (11). TxA2 is a vasodilator that inhibits platelet platelet agonist, whereas PGI2 function and counteracts the prothrombotic effects of TxA2. In a recent paper, Cheng and colleagues provide strong experimental evidence for this hypothesis (12). First, they elegantly verified production COX-1 as the dominant enzyme responsible for TxA2 production by using and COX-2 as the dominant source of PGI2 PGHS-2 knockout (KO) mice; mice in which the COX-2 was inactivated; selective COX-2 inhibition by celecoxib or 5,5-dimethyl-3-(3-fluorophenyl)-4-(4-methylsulphonyl)phenyl-2(5H)-furanone (DFU), a potent and selective COX-2 inhibitor; PGHS-1 KO mice; and PGHS-1 “knockdown” mice (whose expression of PGHS-1 is greatly reduced, resulting in ~95% inhibition of platelet Tx formation, similar in effect to low-dose aspirin treatment). Second, they signaling is responsible for a demonstrate that disruption of PGI2 thrombogenic response following the application of prothrombotic stimuli [i.e., intravenous administration of collagen or the Tx receptor (TP) agonist U46619], and a similar effect was observed with COX-2 inhibition. In addition to creating a prothrombotic state, elevated blood pressure, which has been reported in patients receiving both tNSAIDs (13) or selective COX-2 inhibitors (14), can contribute to increased cardiovascular risk. Indeed, a strong positive relationship exists between increased blood pressure and increased likelihood of cardiovascular events (such as heart attack, stroke, or cardiac arrest) (15). The inhibition of constitutive COX-2 expression in the kidney results in reduced glomerular filtration rate and increased salt and water retention, both of which promote elevated blood pressure (16). Cheng et al. also demonstrate that PGHS-2 KO, PGHS-2 mutation (causing inactivation of COX), and celecoxib treatment of wild-type mice increase systolic blood pressure (12). It is noteworthy that both the prothrombotic effect and blood pressure elevation caused by celecoxib were mitigated in the PGHS-1 knockdown mice because the findings suggest the cardiovascular risk caused by these phenomena would be reduced by concomitant use of low-dose aspirin. Unfortunately, the authors did not administer aspirin to these mice to determine whether this would induce an effect similar to that observed with genetically manipulated PGHS-1 knockdown.
PGE2 is the most abundant product of both COX isoforms. Accordingly, Cheng and colleagues demonstrate that urinary secretion of PGEM (a metabolite of PGE2) was decreased in PGHS-2 KO mice, mice with PGHS-2 mutation, PGHS-1 KO mice, and PGHS-1 into tissues can knockdown mice. Direct delivery of PGE2 are observed elicit inflammation; indeed, higher amounts of PGE2 in inflammatory lesions (such as arthritic joints), and NSAID- synthesis reduces inflammation, dependent inhibition of PGE2 is an important inflammatory mediator. The indicating that PGE2 concerns about cardiovascular risk with coxibs has aroused interest synthesis, downstream of COX, in the selective targeting of PGE2 which may provide a more beneficial cardiovascular risk profile. is complex The regulation of the conversion of PGH2 into PGE2 and requires the participation of several cytosolic and microsomal synthase synthases. Among these enzymes are microsomal PGE2 (mPGES) -1 and mPGES-2. mPGEs-2 is ubiquitously expressed in diverse tissues (17), whereas mPGES-1 expression is induced after exposure to inflammatory stimuli (18). Immunohistochemical analyses of synovial lining cells in RA patients reveal that mPGES-1 is most abundant in patients with active RA. In contrast, mPGES-2 is present in synovial lining cells from both active and quiescent RA patients, suggesting that mPGES-2 participates in homeostatic generation (17). Progression of rather than inflammatory PGE2 collagen-induced arthritis and pain perception is reduced in mice deficient in mPGES-1 (19). The phenotype of these arthritic mice is comparable to those of mice treated with a COX-2-selective inhibitor or a tNSAID. Unfortunately, experimental data regarding the cardiovascular safety of mPGES-1 deletion are inconsistent. Blood pressure elevation was not observed in mPGES-1 KO mice in the study of Cheng and colleagues (12). In another study, however, mPGES-1 was thought to contribute to blood pressure homeostasis (20). Also, the thrombotic response following prothrombotic stimuli was unchanged in mPGES-1 KO mice (20) as compared to that reported elsewhere for wild-type (WT) mice (12) . Relative to WT mice, urinary PGIM (a metabolite of PGI2) was increased in mPGES KO mice, in contrast to TXM (a metabolite of TxA2). The authors speculate that rediversion of PGH2 may explain the beneficial PGI synthase for the formation of PGI2 cardiovascular phenotype of these mice (12). An increase in PGI2 production in mPGES-1 KO mice, however, was not observed by others following intraperitoneal injection of acetic acid injection (19)expression following acetic acid injection under , and PGI2 lipopolysaccharide (LPS)-primed conditions (which induces COX-2 expression) is even reduced in mPGES-1 KO mice compared to that in WT mice (21). Furthermore, genetic deletion of mPGES-1 may result in a different phenotype from that arising from incomplete mPGES-1 inhibition.
Targeted deletion of mPGES-1 reduces inflammation and pain perception in mice and may yield reduced cardiovascular hazard compared to selective COX-2 inhibitors. Therefore, we could speculate that selective inhibition of mPGES-1 is an attractive alternative for the coxibs in the treatment of chronic inflammatory diseases and pain-related disorders; however, we should not forget that COX-2-specific inhibitors had originally been developed to reduce gastrointestinal complications. Like COX-2, mPGES-1 expression is induced by proinflammatory stimuli. Accumulating evidence even suggests that mPGES-1 and COX-2 are induced concomitantly and that mPGES-1 preferentially couples with COX-2 activity to increase the production of PGE2, which is seen in inflammation (22, 23). Thus, it is possible that gastrointestinal complications with mPGES-1 inhibition are comparable to those with COX-2 inhibition.
Undoubtedly, mPGES-1 holds great potential as an alternative for tNSAIDs and COX-2 inhibitors in the treatment of inflammatory diseases. Experimental results are inconsistent thus far and additional experiments are needed to more accurately predict effectiveness and side effects of mPGES-1 deletion. Currently, the benefits are based on experimental studies, because specific pharmacological inhibitors of mPGES-1 (and also inhibitors of other synthases) are not available yet. Anti-inflammatory capacity, PGE2 and cardiovascular and gastrointestinal risk all remain to be determined in adequately designed prospective clinical trials in the future.
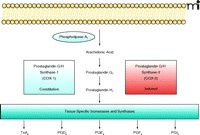
Prostaglandin biosynthesis pathway. Phospholipase A2 catalyzes the release of arachidonic acid (AA) from the membrane, and AA is converted into prostaglandin G2 and H2 by prostaglandin G/H synthase (PGHS)-1 or -2, which contain both cyclooxygenase activity and hydroperoxidase activity. COX-1 is constitutively expressed in many organs, whereas the expression of COX-2 is inducible. Prostaglandin H2 is further metabolized by tissue specific isomerases and synthases [such as PGE synthases (e.g., microsomal PGE synthase-1) or PGI synthase] to generate diverse prostaglandins. Tx, thromboxane; PG, prostaglandin.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Dominique de Kleijn, PhD, is associate professor of Experimental Cardiology at the University Medical Center Utrecht, the Netherlands, and project leader of the Interuniversity Cardiology Institute of the Netherlands. His main interests involve the biology of vascular disease especially plaque destabilization and the response of the heart to myocardial infarction. In these disease entities, our focus has been on inflammation (Toll-like receptors) and we have recently begun to utilize proteomics and lipidomics approaches. E-mail d.dekleijn{at}umcutrecht.nl; fax 0031-30-2522693.

Gerard Pasterkamp, MD, PhD, is head of the laboratory of Experimental Cardiology at the University Medical Center Utrecht. He is working in the research field of arterial remodeling and destabilization of the atherosclerotic plaque. The laboratory of Experimental Cardiology possesses much expertise in the study of animal models of cardiovascular disease. He is chairman of the working group of pathogenesis of Atherosclerosis of the European Society of Cardiology and member of the editorial board of the European Journal of Clinical Investigation. E-mail g.pasterkamp{at}umcutrecht.nl; fax 0031-30-2522693.

Leo Timmers, MD, is a PhD student at the laboratory of Experimental Cardiology at the University Medical Center Utrecht. His focus of interest is translational research of remodeling, cardioprotection, and regeneration following myocardial infarction. He received the Dr. E. Dekker Research Fellowship of the Netherlands Heart Foundation in 2005. In January 2008 he will start a residency in cardiology at the University Medical Center Utrecht. E-mail l.timmers{at}umcutrecht.nl; fax 0031-30-2522693.

