|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2011 | Volume
: 17
| Issue : 3 | Page : 226-228 |
| |
Refractory seizures with global developmental delay: A rare cause
PN Vinoth1, Betty Chacko1, J Julius Xavier Scott1, Venkatasai2
1 Department of Pediatrics, Sri Ramachandra Medical College, Chennai, India
2 Department of Radiology, Sri Ramachandra Medical College, Chennai, India
| Date of Web Publication | 20-Jan-2012 |
Correspondence Address:
J Julius Xavier Scott
Department of Pediatrics, Sri Ramachandra Medical College, Porur, Chennai - 600 116
India
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.92099

 Abstract Abstract | | |
Aicardi syndrome is a genetic disorder characterized by the triad of infantile spasm in flexion, callosal agenesis and ocular abnormalities (chorioretinal lacunae, coloboma of optic disc). We report a typical case of Aicardi syndrome with all the classical features.
Keywords: Callosal agenesis, chorioretinal lacunae, coloboma, infantile spasm
How to cite this article:
Vinoth P N, Chacko B, Xavier Scott J J, Venkatasai. Refractory seizures with global developmental delay: A rare cause. Indian J Hum Genet 2011;17:226-8 |
How to cite this URL:
Vinoth P N, Chacko B, Xavier Scott J J, Venkatasai. Refractory seizures with global developmental delay: A rare cause. Indian J Hum Genet [serial online] 2011 [cited 2016 May 13];17:226-8. Available from: http://www.ijhg.com/text.asp?2011/17/3/226/92099 |
| Introduction | |  |
Aicardi syndrome is a rare genetic disorder characterized by the partial or complete absence of corpus callosum. Triad of spasm in flexion, ocular abnormalities and callosal agenesis were the components of this syndrome described initially by Aicardi et al. [1] Now, skeletal deformities like vertebral anomalies, cortical heterotopias in the eye, mental subnormality and electrophysiological and radiological abnormalities characteristic for this syndrome have been added. [2] We report a typical case of Aicardi syndrome with all the classical features.
| Case Report | | |
A 1-year-old girl presented to us with refractory seizures since the age of 2 months. She was born to second degree consanguineous parents by an uncomplicated normal vaginal delivery at term. There were no significant issues in the antenatal period. She was reported to have myoclonic jerks since the age of 2 months, and later developed to refractory generalized tonic clonic convulsions. She did not achieve any of the age-appropriate milestones, including social smile and head control yet. Her examination revealed microcephaly with marked hypotonia and bilateral extensor plantar response. Fundoscopy in the right disc showed coloboma and multiple areas of choroidal lacunae. Left disc showed retino choroidal coloboma (bridging type) and multiple areas of choroidal lacunae. X-ray of the spine showed kyphoscoliosis and multiple hemivertebrae in the lower thoracic region. X-ray chest and skull were normal. EEG showed burst suppression pattern suggestive of diffuse encephalopathic process. Agenesis of corpus callosum [Figure 1], pachygyria [Figure 2] and arachnoid cyst [Figure 3] were observed in the computed tomography scan of the brain. Karyotyping found the 46 XX pattern. Serological tests for intrauterine infections were negative. The child was commenced on anticonvulsants. Multiple anticonvulsants were needed to decrease the frequency of seizures. Developmental rehabilitation was initiated. Prognosis was explained to parents and need for regular follow-up was advised. However, the child was lost to follow-up.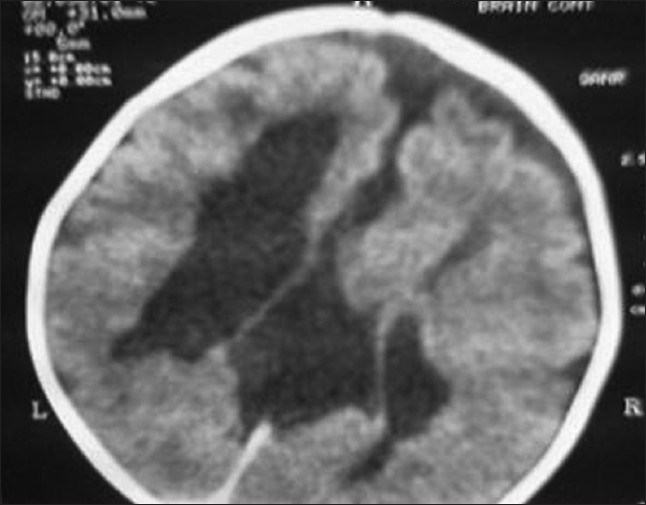 | Figure 1: Computed tomography scan of the brain axial sections show parallel lateral ventricles, agenesis of corpus callosum and asymmetry of cerebral hemispheres
Click here to view |
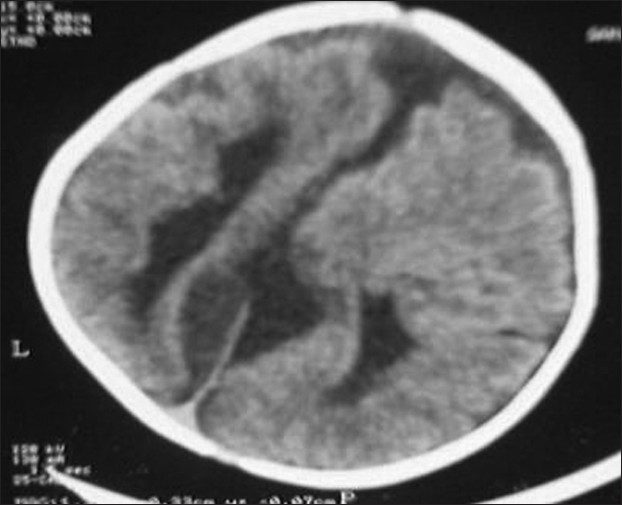 | Figure 2: Computed tomography scan of the brain axial sections show agenesis of corpus callosum, asymmetry of cerebral hemispheres and pachygyria
Click here to view |
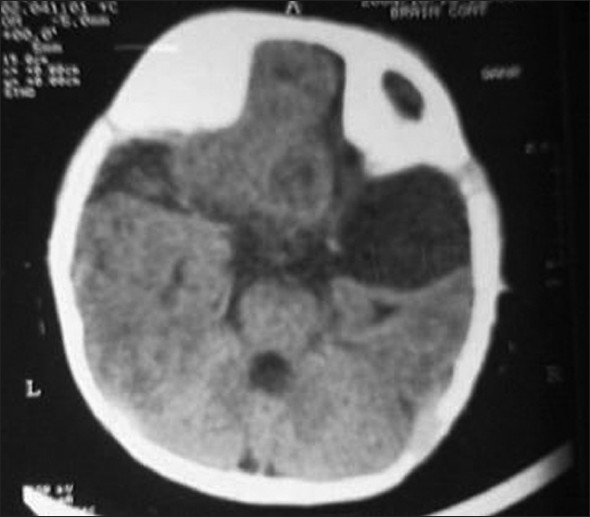 | Figure 3: Computed tomography scan of the brain axial sections show right arachnoid cyst
Click here to view |
| Discussion | | |
Aicardi syndrome is a genetic disorder characterized by the triad of spasm in flexion, callosal agenesis and ocular abnormalities. Aicardi et al., in 1965, first described eight cases with a triad of spasm in flexion, ocular abnormalities and callosal agenesis. [1] Our patient had all the three characteristic features. Now, skeletal deformities like vertebral anomalies, cortical heterotopias in the eye, mental subnormality, electrophysiological and radiological abnormalities characteristic for this syndrome have been added, [2] most of which are present in our case.
X-linked dominant inheritance with mutational event occurring during meiosis in one of the X chromosomes is suggested as a cause. [3] It is mostly lethal in males. The condition has been recognized only in individuals with two X chromosomes; only two were in boys, both with an XXY chromosomal complement. [4] Aicardi syndrome is not a familial condition; only one familial instance is known involving two sisters. [5] The actual frequency of the condition is not known.
Neural tube over distension during embryonic life at 4-8 weeks of gestation is attributed to be the cause for this anomaly. It is often recognized during the neonatal period and infancy, but could be delayed from 11 to 234 weeks after the onset of seizures. [6] Although cases occur throughout the world, the exact incidence and prevalence is unknown.
Seizures in Aicardi syndrome usually start early in life and are mostly myoclonic in type, and other seizures types follow this. Mental retardation, hypotonia and variable pyramidal tracts signs have been observed. Lissencephaly, polygyria, microgyria and cortical heterotopias have been found on pathological examination of the brain. [6]
The most common skeletal abnormality described is fusion of the vertebral bodies. Other abnormalities include hemivertebra, butterfly vertebra, spina bifida occulta, scoliosis, abnormalities in costovertebral articulations, facial and skull asymmetry and increased interorbital distance. [7]
The ophthalmologic findings are usually bilateral, but both eyes are not always equally affected. Opthalmologic findings include coloboma of the optic disc, chorio retinal lacunae, retinal dysplasia with total detachment, micro ophthalmia, persistent pupillary membrane, synechiae of iris and colobomatous cyst. [8] This chorio retinal lacunae has to be differentiated from chorioretinitis of intrauterine infection, especially congenital toxoplasmosis. [9] Bilateral retino choroidal coloboma and choriodal lacunae were present in our child, and the TORCH titers were negative.
Agenesis of corpus collasum and cortical heterotopias are the hallmark of this syndrome. But, asymmetry of the cerebral hemispheres arachnoid cyst, Dandy Walker syndrome, colpocephaly, porencephalic cyst, pachygyria and parallel lateral ventricles have also been described. [10] Our patient had parallel lateral ventricles, arachnoid cyst and pachygyria in addition to collasal agenesis. Microscopic evaluation of the parenchyma commonly reveals disordered cellular organization and disruption of the normal layered appearance of the cortex.
EEG pattern is characteristic, which consists of multifocal epileptiform discharges occurring in the burst suppression pattern, showing complete asynchrony between the two hemispheres. Anatomical and electrophysiological disconnection between the two hemispheres is proposed to be the cause for this EEG abnormality. [11]
The course and outcome of Aicardi syndrome is extremely severe. The condition often leads to death in the first decade. Sudden, unexplained death can occur, but patients commonly die from pulmonary infections.
We report this case to alert pediatricians to suspect this syndrome when they come across girl children with seizures and developmental delay. Careful ophthalmologic examination and electrophysiological and radiological investigations are essential to diagnose this syndrome and to differentiate it from intrauterine infection.
| References | | |
| 1. | Aicardi J, Lefebvre J, Lerique-Koechlin A. A new syndrome: Spasms in flexion, callosal agenesis, ocular abnormalities. Electroencephalogr Clin Neurophysiol 1965;19:609-10. 
|
| 2. | Singhi PD, Gupta A, Agarwal A. Aicardi syndrome. Indian Pediatr 1991;28:1513-6.
[PUBMED] |
| 3. | Ropers HH, Zuffardi O, Bianchi E, Tiepolo L. Agenesis of corpus callosum, ocular and skeletal anamalies (X linked dominant Aicardi's syndrome in a girl with balanced X/3 translocation). Hum Genet 1982;61:364-8.
[PUBMED] |
| 4. | Hopkins IJ, Humphrey J, Keith CG, Sussman M, Webb GC, Turner EK. The Aicardi syndrome in a 47, XXY male. Aust Pediatr J 1979;15:278-80.
|
| 5. | Molina JA, Mateos, F, Merino M, Epistefanio JL, Gorrono M. Aicardi syndrome in two sisters. J Pediatr 1989;115:282-3.
|
| 6. | Bertoni JM, Von Loh S, Allen RJ. The Aicardi syndrome: Report of 4 cases and review of literature. Ann Neurol 1979;5:475-82.
[PUBMED] |
| 7. | De Jong JG, Delleman JW, Houben M, Manschot WA, de Minjer A, Mol J, et al. Agenesis of the corpus callosum, infantile spasms, ocular anomalies (Aicardi's syndrome). Clinical and Pathologic findings. Neurology 1976;26:1152-8.
[PUBMED] |
| 8. | Font RL, Marines HM, Cartwright J Jr, Bausecman SC. Aicardi syndrome: A clinico pathologic case report including electron microscopic observations. Ophthalmology 1991;98:1727-31.
|
| 9. | Willis J, Rosman NP. The Aicardi syndrome versus congenital infections: Diagnostic considerations. J Pediatr 1980;96:235-9.
[PUBMED] |
| 10. | Phillips HE, Carter AP, Kennedy JL Jr, Rosman NP, O'Connor JF. Aicardi's syndrome: Radiological manifestations. Radiology 1978;127:453-5.
[PUBMED] |
| 11. | Fariello RG, Chun RW, Doro JM, Buncic JR, Prichard JS. EEG recognition of Aicardi's syndrome. Arch Neurol 1977;34:563-6.
[PUBMED] [FULLTEXT] |
[Figure 1], [Figure 2], [Figure 3]
|









