|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2012 | Volume
: 18
| Issue : 1 | Page : 130-133 |
| |
A child with mosaicism for deletion (14)(q11.2q13)
Thilini H Gamage1, Imaya U.H. Godapitiya2, Shakila Nanayakkara3, Rohan W Jayasekara1, Vajira H.W. Dissanayake4
1 Human Genetics Unit, University of Colombo, Sri Lanka
2 Asiri Centre for Genomic and Regenerative Medicine, Asiri Surgical Hospital, Colombo, Sri Lanka
3 Neonatal Unit, Castle Street Hospital for Women, Colombo 08, Sri Lanka
4 Human Genetics Unit, University of Colombo; Asiri Centre for Genomic and Regenerative Medicine, Asiri Surgical Hospital, Colombo, Sri Lanka
| Date of Web Publication | 26-May-2012 |
Correspondence Address:
Vajira H.W. Dissanayake
Human Genetics Unit, University of Colombo, Kynsey Road, Colombo 08
Sri Lanka
 Source of Support: None, Conflict of Interest: None  | 3 |
DOI: 10.4103/0971-6866.96684

 Abstract Abstract | | |
In this case report we describe a child with a de novo deletion in the (q11.2q13) region of chromosome 14. The child presented with dysmorphic features - anophthalmia, microcephaly, and growth retardation. Cytogenetic studies showed mosaicism. The karyotype was 46,XX,del(14)(q11.2;q13) [16] /46,XX [9]. We compared the features observed in this child with that of others with the same deletion reported in scientific literature and found that this is the first report of a child mosaic for this deletion. It is also the first time it has been reported in association with anophthalmia.
Keywords: Anophthalmia, deletion (14)(q11.2q13), microcephaly, mosaicism
How to cite this article:
Gamage TH, Godapitiya IU, Nanayakkara S, Jayasekara RW, Dissanayake VH. A child with mosaicism for deletion (14)(q11.2q13). Indian J Hum Genet 2012;18:130-3 |
How to cite this URL:
Gamage TH, Godapitiya IU, Nanayakkara S, Jayasekara RW, Dissanayake VH. A child with mosaicism for deletion (14)(q11.2q13). Indian J Hum Genet [serial online] 2012 [cited 2016 Jun 1];18:130-3. Available from: http://www.ijhg.com/text.asp?2012/18/1/130/96684 |
| Introduction | |  |
Interstitial deletions in the long arm of chromosome 14 are rare. Only a few reports have been published to date reporting deletions in the (14) (q11.2q13) region. [1],[2],[3] Each report has contributed in widening the spectrum of clinical features associated with the deletion. Here, we discuss the first case with this deletion that is present in the mosaic form and associated with anophthalmia.
| Case Report | | |
A seven-month-old baby girl was referred to our centre for genetic evaluation and testing. The child was the first baby born to healthy non-consanguineous parents. She was born at 39 weeks gestation by elective lower section caesarian section following an uneventful pregnancy. At birth her weight, length and occipito frontal circumference (OFC) were 1.325 kg (<3 rd centile), 39 cm (<3 rd centile), and 23 cm (<3 rd centile), respectively.
An ultrasound scan of the brain showed dilation of both lateral ventricles, absence of corpus callosum, asymmetric hypoplasia of the left hemisphere with increased left subarachnoid space. There was no midline shift. The posterior fossa was normal. A 2D echocardiogram showed that cardiac function and structure were normal.
The following features were noted at dysmorphic evaluation: microcephaly, hypotonia, triangular-shaped face, anophthalmia, low set hypoplastic ears, small nose with flat nasal bridge, micrognathia, hypoplastic arms, and puffy hands and feet with high medial arch. The baby weighed 1.8 kg. She had only gained approximately 0.475 kg since birth. The parents declined consent for photographing the child.
The karyotyping was done according to standard protocols of GTL banding technique. The maximum banding resolution achieved was 500 bands. Two cell lines were seen in spreads harvested from both cultures; a cell line showing a deletion with a karyotype of 46,XX,del(14)(q11.2;q13) [Figure 1] and a normal cell line with a karyotype of 46,XX. The parents were karyotyped and found to be normal.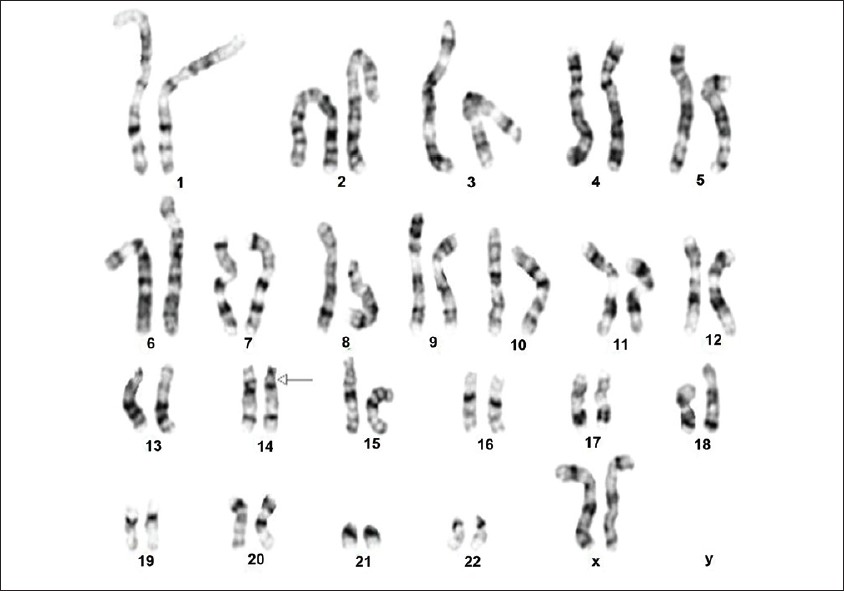 | Figure 1: Karyogram of the cell line showing the deletion (14) (q11.2q13)
Click here to view |
| Discussion | | |
A search of the scientific literature showed that deletion (14)(q11.2q13) was rare. It is associated with a wide spectrum of clinical features with no single distinguishing feature that differentiates it from other structural chromosomal abnormalities. The clinical features of the baby we report above overlap with the features reported in other babies with the same deletion [1],[2],[3] as well as deletions in proximal 14q that involve some of the bands deleted in this baby. [4],[5] These features are compared in [Table 1]. This comparison shows that babies with a deletion confined to 14q11.2 had developmental delay, hypertelorism, short nose with broad flat nasal bridge, prominent philtrum, and muscular hypotonia [5] while babies with the deletion confined to 14q12 had microcephaly, mental retardation, developmental regression, and hypotonia. [4] The baby that we report here had a deletion overlapping both these areas, and hence had a combination of features mentioned above. This baby however differed from all others before by the presence of anophthalmia. | Table 1: Comparison of clinical features in patients with proximal 14q deletions reported in scientific literature
Click here to view |
The loss of the Fork-head box protein G1 (Brain factor-1) gene (FOXG1B), which is mapped to 14q12 [Figure 2], is reported to be responsible for brain malformations, microcephaly, mental retardation. [4],[6] The expression of this gene is restricted to fetal and adult brain and testis. Brain development is assumed to be one of the main functions of this gene. [7] Brain specific expression of this gene explains the severe brain defects and psychomotor retardation observed in our patient and in other patients with FOXG1B gene deletion or disruption. [6]  | Figure 2: An ideogram of chromosome 14 showing the regions deleted in the case reports compared in Table 1. The location of FOXG1B is indicated. The blue bars indicate the regions deleted in the cases compared in Table 1. X is the baby reported in this case report
Click here to view |
A study including a patient with delayed development has shown an instance where an inversion had occurred in chromosome 14 with a balanced translocation where the inversion breakpoint had crossed the FOXG1B gene disrupting the gene. This was postulated to be the most likely cause of mental retardation and microcephaly of the patient. [6] The phenotype of another patient with mental retardation and microcephaly was explained by the deletion of one or more genes in the 14q12 region including the FOXG1B gene. [8]
Absence or agenesis of corpus callosum is another feature present in patients with proximal 14q deletions. [6],[7] This has also been attributed to the loss of the FOXG1B gene. Knockout mouse models of the mouse orthologue of FOXG1B gene have shown that it is necessary for corpus callosum development, [7],[8] which in turn explains the complete agenesis of corpus callosum in this patient.
Anophthalmia associated with abnormalities in chromosome 14 has been reported in cases with 14q22-23 deletions. Two genes, SIX6 and BMP-4, that are responsible for eye development, are present in the 14q22q23 region. [9] A review of scientific literature reporting14q11.2q13 deletions did not yield any previous cases with anophthalmia. This feature may be a novel phenotype associated with the disruption of a yet unidentified genes associated with eye development in this region.
This child was mosaic for two cell lines- one with a normal karyotype 46,XX and one with the deletion 46,XX,del(14)(q11.2;q13). This is unique to our case compared to other cases with the same deletion. Presence of a normal cell line usually reduces the clinical effect of the chromosomal abnormality, but in our case most of the features overlapped with previously published deletions at 14q11.3-q12 were observed along with anophthalmia.
In mosaic cases a normal cell line is, most likely, consistent with a post-zygotic event. A break during DNA replication in the zygote would generate a deleted chromatid, immediately stabilizes by telomeric healing. [10] Segregation gave rise to a cell line with normal karyotype and another with the 14q11.2-q13 deletion [Figure 3].  | Figure 3: Suggested mechanism for the formation of mosaic cell lines in chromosomal deletions
Click here to view |
In conclusion the phenotypic features observed in this baby add to the spectrum of clinical features seen in children with deletions in the proximal 14q region.
| References | | |
| 1. | Grammatico P, de Sanctis S, di Rosa C, Cupilari F, del Porto G. First case of deletion 14q11.2q13: Clinical phenotype. Ann Genet 1994;37:30-2. 
[PUBMED] |
| 2. | Ramelli GP, Remonda L, Lovblad KO, Hirsiger H, Moser H. Abnormal myelination in a patient with deletion 14q11.2q13.1. Pediatr Neurol 2000;23:170-2.
|
| 3. | Schuffenhauer S, Leifheit HJ, Lichtner P, Peters H, Murken J, Emmerich P. De novo deletion (14)(q11.2q13) including PAX9: Clinical and molecular findings. J Med Genet 1999;36:233-6.
[PUBMED] [FULLTEXT] |
| 4. | Mencarelli MA, Kleefstra T, Katzaki E, Papa FT, Cohen M, Pfundt R, et al. 14q12 Microdeletion syndrome and congenital variant of Rett syndrome. Eur J Med Genet 2009;52:148-52.
[PUBMED] [FULLTEXT] |
| 5. | Zahir F, Firth HV, Baross A, Delaney AD, Eydoux P, Gibson WT, et al. Novel deletions of 14q11.2 associated with developmental delay, cognitive impairment and similar minor anomalies in three children. J Med Genet 2007;44:556-61.
[PUBMED] [FULLTEXT] |
| 6. | Shoichet SA, Kunde SA, Viertel P, Schell-Apacik C, von Voss H, Tommerup N, et al. Haploinsufficiency of novel FOXG1B variants in a patient with severe mental retardation, brain malformations and microcephaly. Hum Genet 2005;117:536-44.
[PUBMED] [FULLTEXT] |
| 7. | Ariani F, Hayek G, Rondinella D, Artuso R, Mencarelli MA, Spanhol-Rosseto A, et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet 2008;83:89-93.
[PUBMED] [FULLTEXT] |
| 8. | Bisgaard AM, Kirchhoff M, Tumer Z, Jepsen B, Brondum-Nielsen K, Cohen M, et al. Additional chromosomal abnormalities in patients with a previously detected abnormal karyotype, mental retardation, and dysmorphic features. Am J Med Genet A 2006;140:2180-7.
|
| 9. | Ahmad ME, Dada R, Dada T, Kucheria K. 14q(22) deletion in a familial case of anophthalmia with polydactyly. Am J Med Genet A 2003;120A:117-22.
[PUBMED] [FULLTEXT] |
| 10. | Bonaglia MC, Giorda R, Beri S, Bigoni S, Sensi A, Baroncini A, et al. Mosaic 22q13 deletions: Evidence for concurrent mosaic segmental isodisomy and gene conversion. Eur J Hum Genet 2009;17:426-33.
[PUBMED] [FULLTEXT] |
[Figure 1], [Figure 2], [Figure 3]
[Table 1]
| This article has been cited by | | 1 |
Identical deletion at 14q13.3 including PAX9 and NKX2-1 in siblings from mosaicism of unaffected parent |
|
| Shin Hayashi,Mariko Yagi,Ichijiro Morisaki,Johji Inazawa | | Journal of Human Genetics. 2015; | | [Pubmed] | [DOI] | |
|
|
|
|









