|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2012 | Volume
: 18
| Issue : 3 | Page : 346-348 |
| |
Novel mutation in an Indian patient with Methylmalonic Acidemia, cblA type
Katta Mohan Girisha1, Aroor Shrikiran1, Abdul Mueed Bidchol1, Osamu Sakamoto2, Puthiya Mundyat Gopinath3, Kapaettu Satyamoorthy3
1 Department of Pediatrics, Kasturba Medical College, Manipal University, Manipal, India
2 Tohoku University School of Medicine, Japan
3 Manipal Life Sciences Center, Manipal University, Manipal, Karnataka, India
| Date of Web Publication | 4-Mar-2013 |
Correspondence Address:
Katta Mohan Girisha
Division of Medical Genetics, Department of Pediatrics, Kasturba Medical College, Manipal University, Manipal - 576 104, Karnataka
India
 Source of Support: We thank Technology Information, Forecasting and Assessment Council (TIFAC).Centre of Relevance and Excellence (CORE), Department of Science and Technology, Government of India and Vision Group on Science and Technology (VGST), Department of Information Technology, Biotechnology and Science and Technology, Government of Karnataka for financial support,, Conflict of Interest: None
DOI: 10.4103/0971-6866.108025

 Abstract Abstract | | |
We report on a girl with methylmalonic acidemia, cblA type with a novel homozygous mutation and describe the clinical phenotype and response to therapy.
Keywords: Genetics, methylmalonic acidemia, mutation
How to cite this article:
Girisha KM, Shrikiran A, Bidchol AM, Sakamoto O, Gopinath PM, Satyamoorthy K. Novel mutation in an Indian patient with Methylmalonic Acidemia, cblA type. Indian J Hum Genet 2012;18:346-8 |
How to cite this URL:
Girisha KM, Shrikiran A, Bidchol AM, Sakamoto O, Gopinath PM, Satyamoorthy K. Novel mutation in an Indian patient with Methylmalonic Acidemia, cblA type. Indian J Hum Genet [serial online] 2012 [cited 2016 Jun 1];18:346-8. Available from: http://www.ijhg.com/text.asp?2012/18/3/346/108025 |
| Introduction | |  |
Methylmalonic acidemia refers to a group of organic acidemias characterized by elevation of methylmalonic acid in blood and urine. Isolated methylmalonic acidemia (without homocysteinemia or homocysteinuria) result from complete or partial deficiency of methylmalonyl-CoA mutase (MUT), diminished synthesis of its cofactor 5-deoxyadenosylcobalamin (cblA, cblB, cblD variant-2 complementation groups coded by MMAA, MMAB and MMADHC) and deficiency of methylmalonyl CoA-epimerase (MCEE). We describe an Indian child with methylmalonic acidemia (cblA type, MMAA) (OMIM 251100) describing the natural history and novel mutations in MMAA gene (OMIM 607481).
| Case Report | | |
One-and-a-half year old girl, born to non-consanguineous parents presented with sudden onset of protracted vomiting and unconsciousness of two days duration. She did not have fever. She was apparently asymptomatic till then. She had attained developmental milestones appropriately. She was able to walk independently, scribble, show few body parts and was able to speak a few meaningful words. Her growth was subnormal with both weight (7.4 kg) and length (76 cm) being less than third centile for age. On examination, her Glasgow Coma Scale score was five. She had hypotonia of all four limbs with brisk deep tendon reflexes. Bilateral plantar reflexes were extensors. There were no signs of meningeal irritation. She had a palpable liver 3 cm below costal margin with a span of 7 cm. Other systems were within normal limits. Her initial investigations revealed hemoglobin of 10.3 g%, total leucocyte count of 8300/mm 3 , platelet count of 2,79,000/mm 3 . Neutrophils comprised 84% and lymphocytes 14%. Her blood glucose concentration was 106 mg/dL (60-150 mg/dL), blood urea 42 mg/dl (8-32 mg/dL), serum creatinine 0.4 mg/dL (0.6-1.6 mg/dL), serum sodium 114 mEq/L (130-143 mEq/L), and serum potassium 5 mEq/dL (3.5-5 mEq/L). Liver enzymes were normal with aspartate transaminase of 37 U/L (5-40 U/L), alanine transaminase of 34 U/L (5-40 U/L), and alkaline phosphatase of 197 U/L (40-140 U/L). Cerebrospinal fluid analysis showed no cells on microscopy and gram stain and culture were negative for any organism. CSF glucose was 100 mg/dL (more than or equal to two thirds of blood sugar) and CSF protein was 54 mg/dL (10-20 mg/dL). Arterial blood gas analysis revealed a pH of 7.073 (7.35-7.45), PaO 2 of 214 mmHg, PaCO 2 of 8.2 mmHg (35-45 mmHg), HCO 3 - of 2.3 mmol/L (20-28 mmol/L) suggesting severe metabolic acidosis with a high anion gap. Serum ammonia was 2834 μg% (25-94 μg%) suggesting severe hyperammonemia. Serum lactate was 26 mg/dL (5-22 mg/dL). Thin layer chromatography of urine revealed moderate organic aciduria with significant ketonuria and excretion of valine and alanine. She was treated with intravenous sodium benzoate and fluids. Gradually her sensorium improved and she recovered from the acute illness. However she had residual left lower limb monoparesis. She was started on oral sodium benzoate, thiamine, biotin and carnitine and continued on the same.
Over next six months she gradually improved and achieved normal development except in gross motor domain. By two years of age she was speaking with two word sentences and achieved bowel control. She was able to walk with support. Over the next 4 to 4½ years period, she was regularly followed up for continuation of medications, development assessment, nutritional management and rehabilitation. She had three more acute episodes with vomiting and lethargy that required admissions and intravenous fluids. At 6.5 years of age, she was performing well at school and had good interaction with parents and peers. However she had weakness in the lower limbs and required assistance in walking. At this stage, her urine was subjected to gas chromatography and mass spectrometry, which revealed elevated methylmalonic acid. Tandem mass spectrometry showed significantly elevated propionyl carnitine and normal amino acid profile.
Genomic DNA was extracted from mother and proband. Direct sequencing of all the coding exons including flanking introns of MUT, MMAA and MMAB was carried out as described previously. [1] Alignment of amino acid sequences of methylmalonic aciduria type A protein was carried out by Clustal W ( http://www.ebi.ac.uk/Tools/msa/clustalw2 ) as per the Entrez Protein database. SIFT and Polyphen are in silico tools which are used to predict the effect of amino acids substitutions. SIFT predicts whether an amino acid substitution affects protein function based on sequence homology and the physical properties of amino acids. [2] Polyphen predicts the amino acid substitution based on the structure and function of a human protein using straightforward physical and comparative considerations. [3] The missense mutation was submitted to SIFT ( http://sift.jcvi.org/ ) and PolyPhen ( http://genetics.bwh. harvard.edu/pph/ ).
In exon 6 of MMAA, a G to A substitution at position 833 was found in a homozygous pattern (c.833G > A) [Figure 1]. This mutation leads to the substitution of glycine by aspartic acid at position 278 (p.G278D). The mother showed the same mutation in a heterozygous pattern.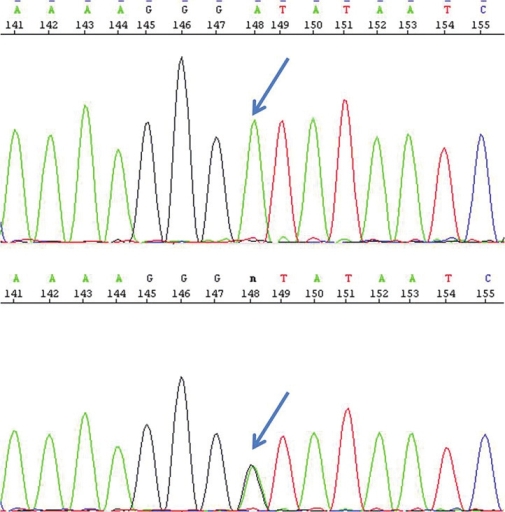 | Figure 1: Electropherogram showing homozygosity (top) in the child and heterozygosity (bottom) in the mother for the mutation c.833G>A indicated by arrow
Click here to view |
Effects of missense mutation were predicted by SIFT and Polyphen. According to SIFT tool, the substitution at position 278 from G to D is predicted to affect protein function with a score of 0.02 (amino acids with probabilities <0.05 are predicted to be deleterious). According to the Polyphen, this variant is predicted to be probably damaging. Alignment of amino acids sequence of methylmalonic aciduria type A protein observed in the patient (resulting in a change of glycine to aspartic acid residue at position 278) showed it to be highly conserved across other species [Figure 2].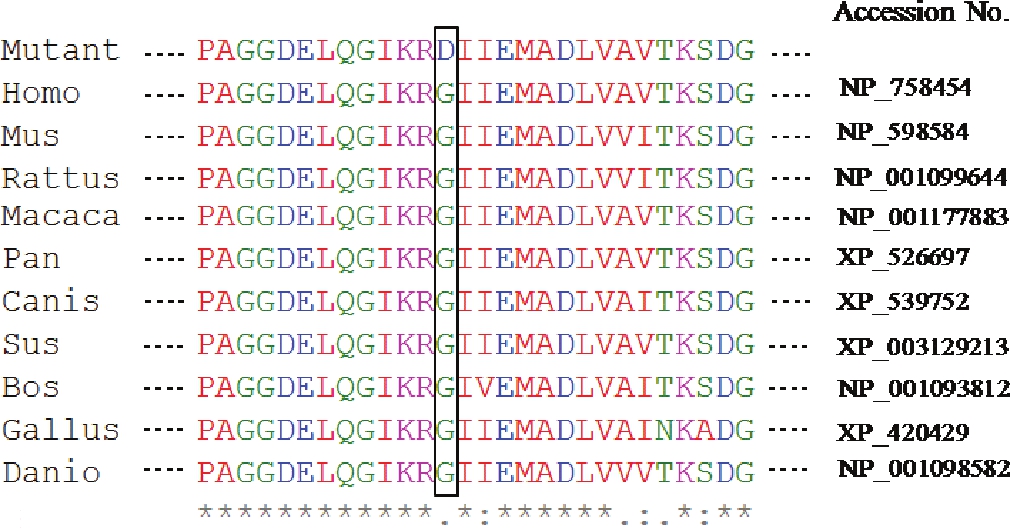 | Figure 2: Comparison with sequences of lower animals shows that the mutation is in the highly conserved site of the gene
Click here to view |
Presently she is on daily oral supplementation of vitamin B12 one milligram and carnitine 500 mg. She is now 7 years and has normal intellectual functions and residual weakness in left lower limb.
During her mother's subsequent pregnancy, prenatal diagnosis was done at 12 weeks of gestation. Same mutation was confirmed in the fetus in homozygous state and she discontinued the pregnancy.
| Discussion | | |
We describe an Indian child with methylmalonic acidemia cblA type. We report the natural history and outcome during a period of follow up of more than five years. We identified a mutation in homozygous state in the proband and heterozygosity for the same mutation in mother. To the best of our knowledge, the mutation described in this patient has not been reported earlier in literature and databases (Human Genome Mutation Database). Identifying the mutation in patients with an inborn error of metabolism has helped this family in early prenatal diagnosis by chorionic villi sampling in the subsequent pregnancy for the family.
Limited number of reports on Indian patients with methylmalonic acidemia is available. [4],[5] However we could find only one earlier report on mutation analysis of methylmalonic acidemia cblC type. [6] We did not find any mutation reported in Indian patients with methylmalonic acidemia cblA type in the literature. We describe here a novel c.833G > A mutation in an Indian family with methylmalonic acidemia type cblA and prenatal diagnosis. We also give an account of phenotype of the patient.
| Acknowledgements | | |
We are grateful to the patient and her family for taking part in this study. We thank Technology Information, Forecasting and Assessment Council (TIFAC)-Centre of Relevance and Excellence (CORE), Department of Science and Technology, Government of India and Vision Group on Science and Technology (VGST), Department of Information Technology, Biotechnology and Science and Technology, Government of Karnataka for financial support.
| References | | |
| 1. | Yang X, Sakamoto O, Matsubara Y, Kure S, Suzuki Y, Aoki Y, et al. Mutation analysis of the MMAA and MMAB genes in Japanese patients with vitamin B 12 responsive methylmalonic acidemia: Identification of a prevalent MMAA mutation. Mol Genet Metab 2004;82:329-33. 
[PUBMED] |
| 2. | Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812-4.
[PUBMED] |
| 3. | Sunyaev S, Ramensky V, Koch I, Lathe W 3 rd , Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet 2001;10:591-7.
|
| 4. | Muranjan MN, Kondurkar P. Clinical features of organic acidemias: Experience at a tertiary care center in Mumbai. Indian Pediatr 2001;38:518-24.
[PUBMED] |
| 5. | Singhi P, Aulakh R, Singh P. Infantile spasms, cerebral venous thrombosis and acute extrapyramidal symptoms in a child with methylmalonic acidemia. J Pediatr Neurol 2009;7:203-7.
|
| 6. | Puri D, Bijarnia S, Thakur S, Verma IC. Methylmalonic Aciduria and Homocystinuria, Cobalamin C (cblC) type. Genet Clin 2009;2:10-1.
|
[Figure 1], [Figure 2]
|









