|
 
 |
|
ORIGINAL ARTICLE |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 1 | Page : 18-25 |
| |
Investigation of cytocrom c oxidase gene subunits expression on the Multiple sclerosis
Naeimeh Safavizadeh1, Seyed Ali Rahmani1, Mohamad Zaefizadeh2
1 Department of Biology, Ahar Branch, IAU university, Ahar, Iran
2 Department of Biology, Ardabil Branch, IAU university, Ardabil, Iran
| Date of Web Publication | 4-Jun-2013 |
Correspondence Address:
Mohamad Zaefizadeh
Department of Biology, Ardabil Branch, IAU university, P.O. Box: 464 Ardabil
Iran
 Source of Support: None, Conflict of Interest: None  | 5 |
DOI: 10.4103/0971-6866.112879

 Abstract Abstract | | |
Introduction: Multiple sclerosis (MS) is an autoimmune inflamatory disease, which affects the (Central Nervous System) and leads to the destruction of myelin and atrophy of the axons. Genetic factors, in addition to environmental ones, seem to play a role in MS. Numerous studies have reported mitochondrial defects including a reduction in cytochrome c oxidase (COX) complex function related to the reduction of mitochondrial genes expression in the cortex tissue of patients with MS have been reported.
Materials and Methods: This study aimed to assess COX5B and COX2 genes expression in MS patients and compare it with normal subjects. We determine expression levels of genes COX5B and COX2, and also gene reference ß-actine using real-time polymerase chain reaction (RT-PCR) method. Data were obtained and obtained and standardized with the gene reference and were analyzed using independent sample t-test with SPSS and Excel programs.
Result and Discussion: The resultshowed COX5B gene expression reduced signifi cant in MS patients compared to normal subjects (P < -0.05) whereas, there was no significant difference in the COX2 gene expression between normal subjects and patients. Thus, it can be claimed that down-regulation of mitochondrial electron transport chain genes supported the hypothesis that hypoxia-like tissue injury in MS may be due to mitochondrial genes, different expression impairment.
Keywords: Cytochrome c oxidase complex, COX2, COX5B, mitochondria, multiple sclerosis, RT-PCR
How to cite this article:
Safavizadeh N, Rahmani SA, Zaefizadeh M. Investigation of cytocrom c oxidase gene subunits expression on the Multiple sclerosis. Indian J Hum Genet 2013;19:18-25 |
How to cite this URL:
Safavizadeh N, Rahmani SA, Zaefizadeh M. Investigation of cytocrom c oxidase gene subunits expression on the Multiple sclerosis. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:18-25. Available from: http://www.ijhg.com/text.asp?2013/19/1/18/112879 |
| Introduction | |  |
Multiple sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system with axonal degeneration. [1] The loss of myelin in MS may be the result of direct damage to myelin through immune mediated processes and dysfunction of oligodendrocytes. [2] Approximately, 15-20% of MS patients have a family history of MS, however, large extended pedigrees are uncommon, with most MS families having no more than two or three affected individuals. Studies in twins [3] and conjugal pairs indicate that much of this familial clustering was the result of shared genetic risk factors, while studies of migrants [4] and apparent epidemics [5] indicated a clear role for environmental factors. Mitochondrial defects are known to occur in aging, cancer, heart disease, and a wide variety of degenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), (MS), and Huntington's disease. [6] Mitochondria contain the respiratory chain where energy in the form of adenosine triphosphate (ATP) is most efficiently produced. [7] The mitochondrial respiratory chain is located in the inner mitochondrial membrane and consists of four complexes (complexes I-IV) whilst complex V is directly involved in ATP synthesis. [8] The complexes of the mitochondrial respiratory chain are include of multiple subunits and all but complex II (which is entirely encoded by nuclear DNA) contain proteins encoded by nuclear and mitochondrial DNA (mtDNA). [9] The final respiratory chain complex (complex IV or cytochrome c oxidase [COX]) is the site at which over 90% of oxygen is consumed. This complex is also involved in proton pumping, essential for ATP synthesis. [8] The mammalian COX is composed of 13 subunits of which the three largest are encoded by the mtDNA and form the catalytic core of the enzyme. The remaining ten, evolutionary younger, nuclearly encoded subunits are involved in assembly and regulation of the enzyme. [10] The function of mammalian COX can be physiologically modulated and the enzyme represents one of the key regulatory sites of energy metabolism. [11] COX transfers electrons from cytochrome c to molecular oxygen, which is reduced to water. The electrons pass from cytochrome c, binding at subunit II, through Cu-(A) and heme a cofactors, to the binuclear center buried inside subunit I and composed of heme a3 and Cu-(B), where the incoming four electrons together with four protons from the matrix are sequentially used for oxygen reduction. This exergonic redox reaction is coupled with proton pumping across the inner mitochondrial membrane, but the coupling of the two processes (H + /e - stoichiometry) can be modulated. In addition to Mitchell's chemiosmotic theory, a "second mechanism of respiratory control" has been proposed that involves the binding of adenine nucleotides to nuclear-encoded COX subunits. The key event is the phosphorylation of subunit IV. Activity of phosphorylated COX is regulated by adenosine triphosphate/adenosine diphosphate (ATP/ADP) ratio and respiratory rate is precisely controlled according to the ATP utilization. The membrane potential is kept low (100-150 mV) and COX works at high efficiency of proton translocation (H + /e - =1). The COX biosynthesis and assembly is timely and complicated process involving several rate-limiting steps reflecting the sequential incorporation of the subunits from either the cytosol (nuclearly coded subunits) or from the mitochondrial matrix (subunits I, II, and III). The mainly of COX defects thus, originate from mutations in nuclear genes. [12] Mutations in the genes encoding several COX assembly factors have been described as a frequent cause of mitochondrial diseases and have been assigned with specific clinical symptoms. The dysfunction of COX in these cases is mostly caused by structural changes rather than by the changes in amount of the enzyme. [13] COX subunit II, abbreviated COX II, is the second subunit of COX subunit 2 (CO II) transfers the electrons from cytochrome c to the catalytic subunit 1. It contains two adjacent transmembrane regions in its N-terminus and the major part of the protein is exposed to the periplasmic or to the mitochondrial intermembrane space, respectively. CO II provides the substrate-binding site and contains a copper center called Cu-(A), probably the primary acceptor in COX. An exception is the corresponding subunit of the cbb3-type oxidase which lacks the copper (A) redox-centre. Several bacterial CO II have a C-terminal extension that contains a covalently bound haem c. [14] Subunit Vb of mammalian COX is encoded by a nuclear gene and assembled with the other 12 COX subunits encoded in both mitochondrial and nuclear DNA. This gene to chromosome 2, region cen-q13. [15] There is a common symptom of MS with mitochondrial diseases such as (AD), (PD) and also the point mutation mtDNA can cause damage to the myelin. [16] In this study, we aimed to assess COX5B and COX2 genes expression in MS patients and normal subjects were compared. With due attention to mitochondria of importance in MS disease, the main focus our study was use (RT-PCR) for determine expression levels of genes COX5B and COX2.
| Materials and Methods | | |
Subjects
Thirty individuals' patient (12 male, 18 female) and subject (14 male, 16 female) took part in this study. Written informed consent was obtained from each individual. Peripheral blood samples (5 ml) were obtained from the cubital vein and collected in cell preparation tubes containing an anticoagulant Ethylene diamine tetraacetic acid (EDTA). Peripheral blood mononuclear cells were isolated by EDTA density centrifugation.
RNA extraction
Total RNA was isolated by using a RNeasy kit from Roche (Germany), according to the manufacturer's recommendations (200 μl sample input, 100 μl sample output). The RNA samples were incubated with RNAse-free DNAse I at 37°C for 15 min. RNA samples were purified with a RNeasy kit. Extract total RNA immediately or after storage at −80°C for months. Total RNA quality and quantity were assessed in 2 ways. In the first method, we estimated the RNA concentration by ultraviolet absorbance at 260 nm (1 absorbance unit at 260 nm = 40 ng/μl RNA) and the RNA purity by measuring the ratio of absorbance at 260 nm and 280 nm (1.8 < A 260 /A 280 < 2.1 for pure RNA). Total RNA was run on 1% agarose gels to check size and integrity. The quality of RNA was confirmed by the detection of 18S and 28S bands after 1% agarose gel electrophoresis.
Primer synthesis
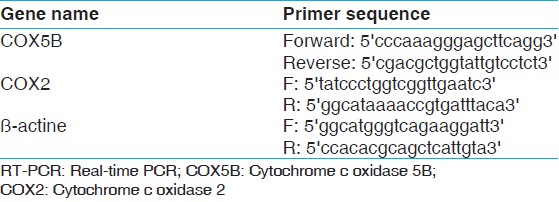
Primer sequences were designed using Primer3 software for COX5B (NM_001862.2), COX2 (NC_012920.1) and ί-actine (reference gene) genes, and then primer sequences edited with the Bioedit software, the primer sequences confirmed in the NCBI/BLAST database [Table 1].
Complementary DNA (cDNA) synthesis
Total RNA from each sample was used to generate cDNA with a Reverse Transcriptase cDNA synthesis kit (Roche, Germany) with oligo deoxythymine (dT) primers, according to the manufacturer's protocol. Briefly, we added 1 μL of oligo (dT), The reagents RNA, oligo (dT) were mixed and then heated at 70°C for 5 min. They were chilled on ice until the other components were added. Then, we added 2 μL deoxyribonucleotid triphosphate (dNTP), 4 μL of buffer, and 1 μL of ribolack (RNase inhibition). The samples were mixed and incubated 37°C for 5 min. Then 1 μL of Reverse Transcriptase was added, and the samples were mixed and incubated at 42°C for 60 min. The reaction was inactivated at 70°C for 10 min. Finally, we cDNA stored at −20°C.
Quantitative RT-PCR
Amplification was performed over 30 cycles on a MJ Research PTC-200 Gradient Cycler (MJ research, Waltham Mass, USA). The annealing temperature was 54°C, extension occurred at 72°C, and denaturation occurred at 94°C. After completion of PCR, the product was separated by electrophoresis in 1% agarose gel and detected by ethidium bromide staining. Only cDNA testing positive for é-actine expression was used in the RT-PCR. Relative expression of COX2, COX5B in the blood samples was carried out by RT-PCR analysis using the ABI PRISM 7700 Sequence Detection System (Applied Biosystems, Darmastadt, Germany) and the Sybr Green PCR Kit (Qiagene). The data were evaluated by the ΔΔCT method, which measures the expression level of the target genes normalized to a reference gene and relative to the expression of the genes in a calibrator samples. After efficiency testing, é-actine was chosen as the reference housekeeping gene and the same primer pairs used for the standard PCR were utilized for the RT-PCR. Amplification was performed over 45 cycles. The annealing temperature was 54°C, extension occurred at 70°C, and denaturation occurred at 94°C. Every cDNA samples were measured in four separate preparations to correct minor variations. Melting curve analyses were carried out after completion to confirm the presence of single amplified species. The transcript levels of COX2, COX5B in the samples were normalized to the transcriptional level of the housekeeping gene é-actine and calculated relative to the expression levels of the target genes in a calibrator blood samples. The arithmetic mean of the relative expression levels of COX2, COX5B for each group of patients and normal subjects were calculated.
Statistical analysis
Statistically significant differences were assessed by the student's t-test using the software's SPSS and Excel. A P < -0.05 was accepted as the level of significance.
| Results | | |
To determine the role of mitochondrial-encoded gene and nuclear-encoded gene in MS pathogenesis, using quantitative RT-PCR analysis with SYBR-Green fluorescent dye, we calculated the mRNA fold change for one mitochondrial-encoded gene in complex IV (COX2) and one nuclear-encoded gene in complex IV (COX5B) of oxidative phosphorylation.
RT-PCR data analysis for genes expression
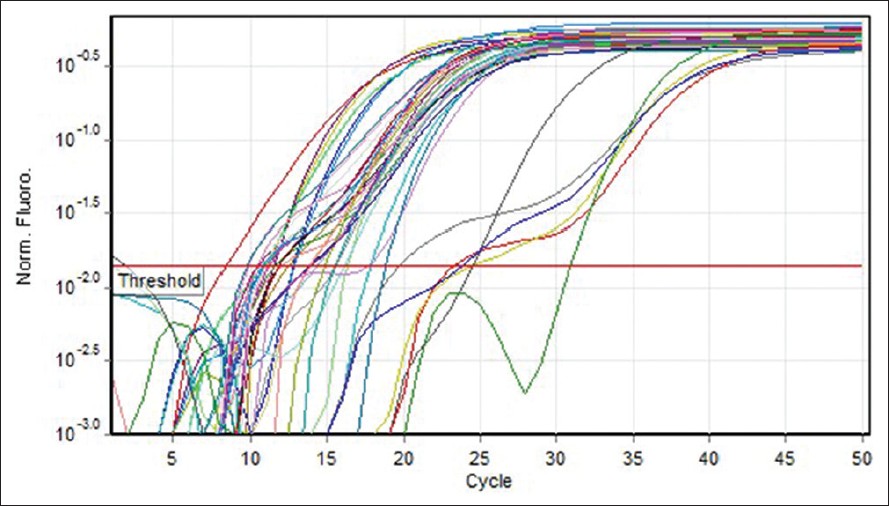
Quantitative measurement of (COX2, COX5B) genes expression was investigated in 30 MS patients (12 males and 18 females). Five different standard concentrations were used to draw a standard curve [Figure 1]. Quantitative measurement of genes expression achieved in the PCR of the cycles was carried out in progressive phase. RT-PCR Software can determine Threshold Cycle automatically [Figure 2]. Furthermore, due to temperature changes in melting curve analysis. There is only one peak associated with the COX5B and COX2 genes, which is a symptom of lack of cases such as non-specific products and primer dimer [Figure 3]. After the PCR, as a template standard DNA of successive dilutions, standard curve was plotted for the COX5B and COX2 genes [Figure 4]. Standard curve of COX5B gene obtained determination of index (R 2 ) = 0.98 and also standard curve COX2 gene obtained detremination of index (R 2 ) = 0.95.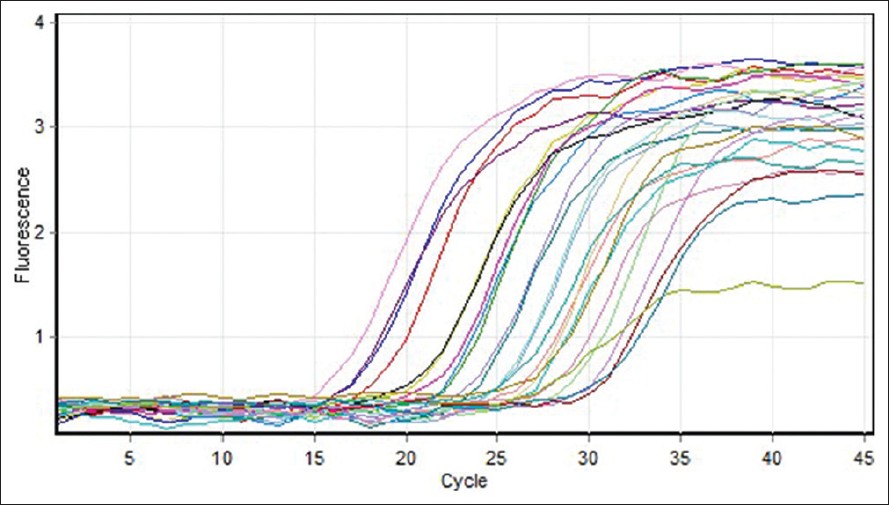 | Figure 1: Gene amplification curves for multiple sclerosis cytochrome c oxidase 5B with real-time polymerase chain reaction
Click here to view |
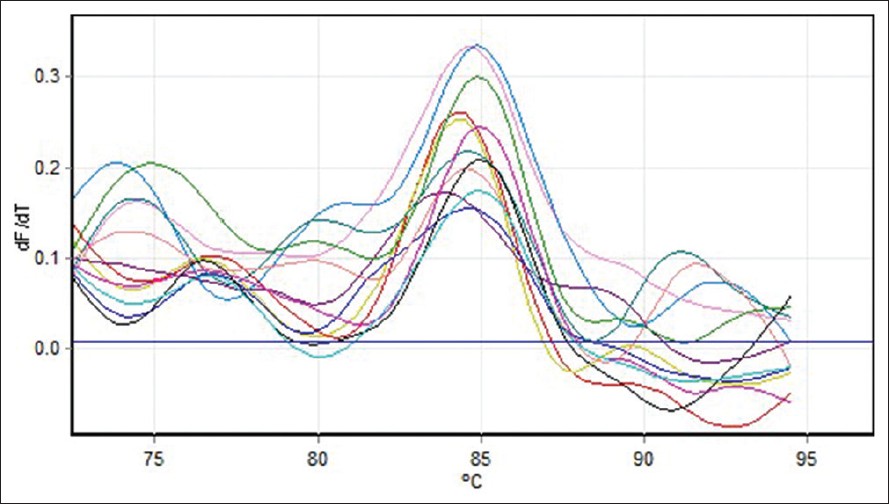 | Figure 3: Melting curve for cytochrome c oxidase 5B gene on the real-time PCR
Click here to view |
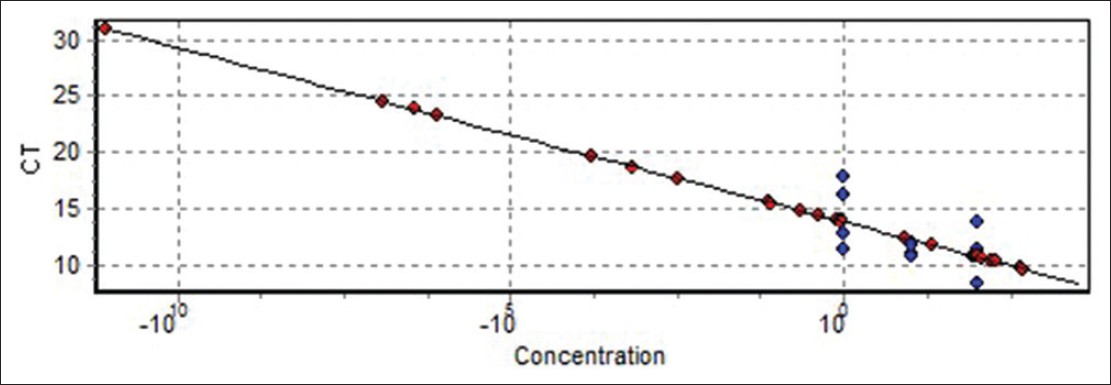 | Figure 4: Standard curve cytochrome c oxidase 5B gene (determination of index R2 = 0.98)
Click here to view |
Standardized results ΔCt
After RT-PCR, studies showed that the ΔCt standard of gene expression β-actine gene COX5B is for MS patients 6.87 and for normal subjects 6.18 and also the -actine gene COX5B is for MS patients 6.87 and for normal subjects 6.18 and also the ΔCt standard of gene expression β-actine gene COX2 is for MS patients 2.79 and for normal subjects 5.24 [Figure 5]. Comparison was performed of the COX5B gene statistical analysis t-test, and the results showed that there are meaning differences in COX5B gene (P < -0.05). And decreased gene expression COX5B was observed in MS patients comparison with normal subjects; and the results showed that there are not meaning differences in COX2 gene [Table 2] and [Table 3]. To continue t-test was performed levene's test for equality of variances. The results showed that the data related to ΔCt were not significant difference for COX5B gene in patiene subjects and normal subjects. Compare ΔCt was performed with t-test in patiene subjects and normal subjects, the results showed that measure t is significant for COX5B gene (P < -0.024). However, the results showed that the data related to ΔCt were significant difference for COX2 gene in patiene subjects and normal subjects. Compare ΔCt was performed with t-test in in patiene subjects and normal subjects, the results showed that measure t is not significant for COX2 gene (P < -0.11) [Table 4] and [Table 5].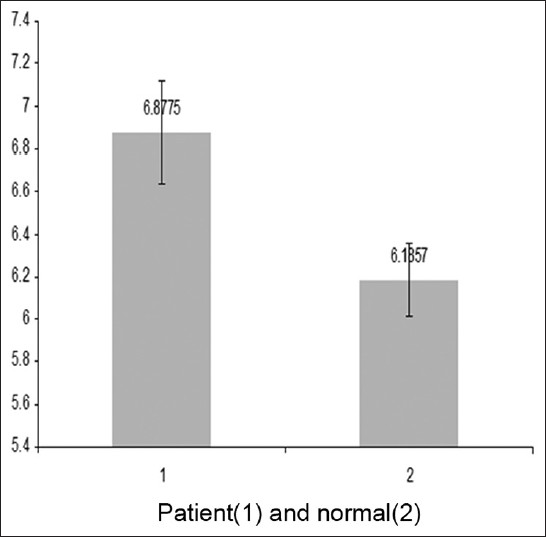 | Figure 5: The Δ Ct cytochrome c oxidase 5B gene that were standardized with reference gene (β-actin)
Click here to view |
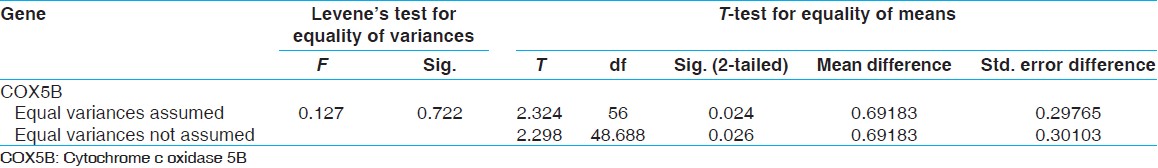 | Table 4: Results t-test for COX5B gene expression on the control and patient
Click here to view |
 | Table 5: Results t-test for COX5B gene expression on the control and patient
Click here to view |
Determine changes in gene expression levels
To determine changes in gene expression COX5B and COX2 after RT-PCR was used livak formula. [17]
COX5B = 2 -ΔΔCT = 2 -(6.18-6.87) =2 0.69 = 1.61
COX2 = 2 -ΔΔCT = 2 -(5.24-2.79) =2 -0.45 = 0.73
Total results of the RT-PCR
According to the results obtained during experiments, it seems that COX2 gene has no effect in MS patients, however, COX5B gene expression was decreased in MS patients and this gene can plays an important role in neurodegenerative diseases. Correlation between COX5B and COX2 genes expression turned out to be R = 0.92 in normal subjects. This correlation was R = 0.84 in patients which, despite being significant, was lower compared to normal subjects. This difference, if verified with further subjects and studies could be considered as a hypothesis about this disease.
| Discussion | | |
In this study, the expression levels of COX5B and COX2 genes in MS patients and normal subjects were compared. The results showed COX5B gene expression in MS patients was significantly lower compared to normal subjects (P < -0.024) while there was no significant difference in the COX2 gene expression between normal subjects and patients, there was not significant different (P < -0.11).
Degeneration of chronically demyelinated axons is a major cause of the continuous, irreversible neurological disability that occurs in the chronic stages of MS. [18] Microarray studies from cortical and white matter tissue of patients with progressive MS showed up-regulation of genes involved in (hypoxic) pre-conditioning and decreased expression of mRNA for mitochondrial proteins. [19] The dominant loss of small caliber axons in multiple sclerosis MS lesions suggests energy deficiency as a major mechanism included in axonal degeneration. [20] The newly reported provide evidence that neurons in MS are respiratory-deficient due to mtDNA deletions, which are extensive in GM and may be induced by inflammation. They propose induced multiple deletions of mtDNA as an important contributor to neurodegeneration in MS. [21] This presents a unique challenge to neurologists wanting to identify, diagnose, and manage patients and families with mitochondrial disease. [22] Clonally expanded multiple deletions of mtDNA causing respiratory deficiency are well recognized in neurodegenerative disorders and aging. Given the vulnerability of mtDNA to oxidative damage and the extent of inflammation in MS, often starting with a preclinical phase and remaining throughout the disease course, mtDNA deletions might be expected in MS. [23] Repair of damaged mtDNA rather than oxidative damage to mtDNA per se is the most likely mechanism by which mtDNA deletions are formed, and clonal expansion is regarded as the mechanism responsible for causing respiratory deficiency. Where respiratory deficiency is caused by induced multiple DNA deletions, cells will have initially contained deletions with different breakpoints, one of which then clonally expands to high levels over time. [24] Clonally expanded multiple deletions of mtDNA are reported in inclusion body myositis, a condition associated with chronic inflammation. Decrease in density of respiratory-deficient neurons in lesions is a likely reflection of mtDNA deletion-mediated cell loss as well as increase in susceptibility to other insults because of the clonally expanded mtDNA deletions. The extent of mtDNA deletions identified using long-range PCR on a global scale regardless of cell type, and even in neurons with intact complex IV activity, reflects the potential of cells in MS to become respiratory deficient through clonal expansion over the course of the disease. By isolating individual neurons at a single time point we identified high levels of multiple mtDNA deletions within respiratory-deficient cells. In contrast, mtDNA deletions detected by long range PCR within respiratory-efficient neurons in MS and controls were not expanded to high levels. [25] Mitochondrial defects are increasingly recognized to play a role in the pathogenesis of MS. Energy in the form of ATP is most efficiently produced by mitochondria, which also play a role in calcium handling, production of reactive oxygen species, and apoptosis. [19] It is known that mitochondria are intrinsically involved in the cellular production of oxygen radicals and are believed to play an important part in oxygen radical-mediated cell damage in neurodegenerative diseases. The investigations are in complete agreement with the occurrence of activity defects of COX in single muscle fibers. The mitochondrial impairment is not to age or denervation-associated muscular changes since the functional impairment of mitochondria. [26] While numerous pathogenetic mutations are routinely detected in isolated COX deficiencies, the protocols for characterizing the functional impact of these mutations are still in early stages of development. The integrative approach combining multiple bioenergetics analyses performed in whole cells is particularly promising, as the best way to investigate the resulting pathogenetic changes occurring in situ. The general severity of the functional changes in COX defects suggests that the development of effective drugs is very unlikely, and that only gene therapy might lead to efficient treatment of these diseases in the future. [27] Measurement of the threshold for specific mtDNA mutations has been performed in cultured cells elsewhere. Themicrophotometric enzyme-assay system for measurement of COX activity within individual muscle fibers is used in conjunction with demonstration of myofibrillar ATPase in serial sections and, hence that a normal range of COX activity in individual muscle fibers of each fiber type is defined in control samples. In previous studies, a good correlation between microphotometric enzyme analysis and biochemical studies has been shown in patients with Leigh syndrome. [28] The newly reported four proteins in particular were responsible for distinguishing disease from control. Peptide fingerprint mapping unambiguously identified these differentially expressed proteins. Three proteins identified are involved in respiration including, COX subunit 5B (COX5B), the brain specific isozyme of creatine kinase, and hemoglobin β-chain. [29] In previous studies, six genes involved in energy metabolism pathways also had increased transcript levels in the skeletal muscle of CR rats compared with muscle of control rats. These include genes associated with mitochondrial ATP production, such as six subunits of COX (COX I, II, III, IV, Va, and VIII) and NADH dehydrogenase. [30] All mitochondria of the progeny are inherited from the mother; and all 13 polypeptides encoded by the mitochondrial genome are located in the respiratory chain (complexes I, III, IV and V). These biological principles are helpful in understanding the clinical syndromes and patterns of inheritance associated with the mitochondrial myopathies and encephalomyopathies. [31] Cellular injury often has been associated with disturbances in mitochondrial function. Interestingly, our microarray analysis indicated a pronounced increase in the expression of mitochondrial COX subunits IV and Vb, a finding, which was validated further by RT-PCR. COX has been used frequently as a marker for neuronal metabolic activity, especially, in pathological conditions involving oxidative stress. Neurons subject to oxidative damage show abnormalities in mitochondrial dynamics even in the absence of any apparent indication of degeneration. [32] Elevations in cytochrome c oxidase expression or function in vulnerable neuronal subpopulations in AD, following traumatic brain injury and preceding apoptotic death of spinal cord motor neurons after sciatic nerve avulsion, have been reported. Thus, an increase in cytochrome c oxidase expression may reflect aberrant energy metabolism and oxidative damage in the spinal cord during EAE. [33]
With due attention to the results correlation between COX2, COX5B genes expressions with nuclear DNA and mtDNA source, We can claim that there are interaction between two COX genes expression of genome and Mitochondria source and the effectiveness is of COX enzymes in the MS patients.
| References | | |
| 1. | Frohman EM, Racke MK, Raine CS. Multiple sclerosis - The plaque and its pathogenesis. N Engl J Med 2006;354:942-55. 
|
| 2. | Vercellino M, Romagnolo A, Mattioda A, Masera S, Piacentino C, Merola A, et al. Multiple sclerosis relapses: A multivariable analysis of residual disability determinants. Acta Neurol Scand 2009;119:126-30.
|
| 3. | Calabresi P. Multiple sclerosis and demyelinating conditions of the central nervous system. In: Goldman L, Ausiello D, editors. Cecil Medicine. 23 rd ed. Ch. 436. Philadelphia, Pa: Saunders Elsevier; 2007.
|
| 4. | Ramagopalan SV, Dobson R, Meier UC, Giovannoni G. Multiple sclerosis: Risk factors, prodromes, and potential causal pathways. Lancet Neurol 2010;9:727-39.
|
| 5. | Koch M, Uyttenboogaart M, van Harten A, De Keyser J. Factors associated with the risk of secondary progression in multiple sclerosis. Mult Scler 2008;14:799-803.
|
| 6. | Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol 2008;4:600-9.
|
| 7. | Damiano M, Galvan L, Déglon N, Brouillet E. Mitochondria in Huntington's disease. Biochim Biophys Acta 2010;1802:52-61.
|
| 8. | Alston CL, He L, Morris AA, Hughes I, de Goede C, Turnbull DM, et al. Maternally inherited mitochondrial DNA disease in consanguineous families. Eur J Hum Genet 2011;19:1226-9.
|
| 9. | Gallardo ME, Moreno-Loshuertos R, López C, Casqueiro M, Silva J, Bonilla F, et al. m. 6267G > A: A recurrent mutation in the human mitochondrial DNA that reduces cytochrome c oxidase activity and is associated with tumors. Hum Mutat 2006;27:575-82.
|
| 10. | Zee JM, Glerum DM. Defects in cytochrome oxidase assembly in humans: Lessons from yeast. Biochem Cell Biol 2006;84:859-69.
|
| 11. | Fernández-Vizarra E, Tiranti V, Zeviani M. Assembly of the oxidative phosphorylation system in humans: What we have learned by studying its defects. Biochim Biophys Acta 2009;1793:200-11.
|
| 12. | Acin-Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab 2009;9:265-76.
|
| 13. | Galati D, Srinivasan S, Raza H, Prabu SK, Hardy M, Chandran K, et al. Role of nuclear-encoded subunit Vb in the assembly and stability of cytochrome c oxidase complex: Implications in mitochondrial dysfunction and ROS production. Biochem J 2009;420:439-49.
|
| 14. | Barrientos A, Gouget K, Horn D, Soto IC, Fontanesi F. Suppression mechanisms of COX assembly defects in yeast and human: Insights into the COX assembly process. Biochim Biophys Acta 2009;1793:97-107.
|
| 15. | Williams JC, Sue C, Banting GS, Yang H, Glerum DM, Hendrickson WA, et al. Crystal structure of human SCO1: Implications for redox signaling by a mitochondrial cytochrome c oxidase assembly protein. J Biol Chem 2005;280:15202-11.
|
| 16. | DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci 2008;31:91-123.
|
| 17. | Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods 2001;25:402-8.
|
| 18. | Aschrafi A, Schwechter AD, Mameza MG, Natera-Naranjo O, Gioio AE, Kaplan BB. MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons. J Neurosci 2008;28:12581-90.
|
| 19. | Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006;443:787-95.
|
| 20. | Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci 2005;233:3-13.
|
| 21. | Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O, et al. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol 2011;69:481-92.
|
| 22. | Druzhyna NM, Wilson GL, LeDoux SP. Mitochondrial DNA repair in aging and disease. Mech Ageing Dev 2008;129:383-90.
|
| 23. | Nicholas A, Kraytsberg Y, Guo X, Khrapko K. On the timing and the extent of clonal expansion of mtDNA deletions: Evidence from single-molecule PCR. Exp Neurol 2009;218:316-9.
|
| 24. | Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, et al. What causes mitochondrial DNA deletions in human cells? Nat Genet 2008;40:275-9.
|
| 25. | Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, et al. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 2006;439:988-92.
|
| 26. | Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol 2006;59:478-89.
|
| 27. | Smith D, Gray J, Mitchell L, Antholine WE, Hosler JP. Assembly of cytochrome-c oxidase in the absence of assembly protein Surf1p leads to loss of the active site heme. J Biol Chem 2005;280:17652-6.
|
| 28. | Ogbi M, Johnson JA. Protein kinase Cepsilon interacts with cytochrome c oxidase subunit IV and enhances cytochrome c oxidase activity in neonatal cardiac myocyte preconditioning. Biochem J 2006;393:191-9.
|
| 29. | Horváth R, Schoser BG, Müller-Höcker J, Völpel M, Jaksch M, Lochmüller H. Mutations in mtDNA-encoded cytochrome c oxidase subunit genes causing isolated myopathy or severe encephalomyopathy. Neuromuscul Disord 2005;15:851-7.
|
| 30. | Kalman B, Laitinen K, Komoly S. The involvement of mitochondria in the pathogenesis of multiple sclerosis. J Neuroimmunol 2007;188:1-12.
|
| 31. | Broadwater L, Pandit A, Clements R, Azzam S, Vadnal J, Sulak M, et al. Analysis of the mitochondrial proteome in multiple sclerosis cortex. Biochim Biophys Acta 2011;1812:630-41.
|
| 32. | Hamblet NS, Ragland B, Ali M, Conyers B, Castora FJ. Mutations in mitochondrial-encoded cytochrome c oxidase subunits I, II, and III genes detected in Alzheimer's disease using single-strand conformation polymorphism. Electrophoresis 2006;27:398-408.
|
| 33. | Fukui H, Diaz F, Garcia S, Moraes CT. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 2007;104:14163-8.
|
[Figure 1], [Figure 2], [Figure 3], [Figure 4], [Figure 5]
[Table 1], [Table 2], [Table 3], [Table 4], [Table 5]
| This article has been cited by | | 1 |
Perturbed Glucose Metabolism: Insights into Multiple Sclerosis Pathogenesis |
|
| Deepali Mathur,Gerardo López-Rodas,Bonaventura Casanova,Maria Burgal Marti | | Frontiers in Neurology. 2014; 5 | | [Pubmed] | [DOI] | |
|
|
|
|