|
 
 |
|
ORIGINAL ARTICLE |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 2 | Page : 171-178 |
| |
Subtelomeric chromosomal rearrangements in a large cohort of unexplained intellectually disabled individuals in Indonesia: A clinical and molecular study
Farmaditya E. P. Mundhofir1, Willy M Nillesen2, Bregje W. M. Van Bon2, Dominique Smeets2, Rolph Pfundt2, Gaby van de Ven-Schobers2, Martina Ruiterkamp-Versteeg2, Tri I Winarni3, Ben C. J. Hamel2, Helger G Yntema2, Sultana M. H. Faradz3
1 Division of Human Genetics, Center for Biomedical Research, Faculty of Medicine Diponegoro University, Semarang, Indonesia; Department of Human Genetics, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands,
2 Department of Human Genetics, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands
3 Division of Human Genetics, Center for Biomedical Research, Faculty of Medicine Diponegoro University, Semarang, Indonesia
| Date of Web Publication | 5-Aug-2013 |
Correspondence Address:
Sultana M. H. Faradz
Division of Human Genetics, Center for Biomedical Research, Faculty of Medicine, Diponegoro University, GSG 2nd Floor, Jl. Dr. Sutomo 14, Semarang
Indonesia
 Source of Support: This research is partly funded by the RISBIN IPTEKDOK
2007/2008 program of the Ministry of Health Republic of Indonesia, Excellent
Scholarship (Beasiswa Unggulan), Overseas study Scholarship (Beasiswa
Luar Negeri) of the Directorate General of Higher Education (DGHE)
Ministry of National Education and Culture Republic of Indonesia and the
PhD.fellowship program of the Radboud University Nijmegen (RU.fellowship), Conflict of Interest: None
PMID: 24019618 
 Abstract Abstract | | |
Context: Unbalanced subtelomeric chromosomal rearrangements are often associated with intellectual disability (ID) and malformation syndromes. The prevalence of such rearrangements has been reported to be 5-9% in ID populations.
Aims: To study the prevalence of subtelomeric rearrangements in the Indonesian ID population.
Materials and Methods: We tested 436 subjects with unexplained ID using multiplex ligation dependent probe amplification (MLPA) using the specific designed sets of probes to detect human subtelomeric chromosomal imbalances (SALSA P070 and P036D). If necessary, abnormal findings were confirmed by other MLPA probe kits, fluorescent in situ hybridization or Single Nucleotide Polymorphism array.
Results: A subtelomeric aberration was identified in 3.7% of patients (16/436). Details on subtelomeric aberrations and confirmation analyses are discussed.
Conclusion: This is the first study describing the presence of subtelomeric rearrangements in individuals with ID in Indonesia. Furthermore, it shows that also in Indonesia such abnormalities are a prime cause of ID and that in developing countries with limited diagnostic services such as Indonesia, it is important and feasible to uncover the genetic etiology in a significant number of cases with ID.
Keywords: Indonesia, intellectual disability, multiplex ligation dependent probe amplification, subtelomeric rearrangements
How to cite this article:
Mundhofir FE, Nillesen WM, Van Bon BW, Smeets D, Pfundt R, de Ven-Schobers Gv, Ruiterkamp-Versteeg M, Winarni TI, Hamel BC, Yntema HG, Faradz SM. Subtelomeric chromosomal rearrangements in a large cohort of unexplained intellectually disabled individuals in Indonesia: A clinical and molecular study. Indian J Hum Genet 2013;19:171-8 |
How to cite this URL:
Mundhofir FE, Nillesen WM, Van Bon BW, Smeets D, Pfundt R, de Ven-Schobers Gv, Ruiterkamp-Versteeg M, Winarni TI, Hamel BC, Yntema HG, Faradz SM. Subtelomeric chromosomal rearrangements in a large cohort of unexplained intellectually disabled individuals in Indonesia: A clinical and molecular study. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:171-8. Available from: http://www.ijhg.com/text.asp?2013/19/2/171/116118 |
FNx01Equal last author
| Introduction | |  |
The genetic causes of intellectual disability (ID) comprise (sub) microscopically chromosome abnormalities and monogenic diseases. [1] Microscopically visible numerical and structural abnormalities are the most common cause of ID. In a large meta-analysis review, a median rate of 9.5% was described. [2] Apart from these microscopically visible chromosomal abnormalities, there are submicroscopic abnormalities that cannot be detected by conventional karyotyping. Abnormalities in the most distal ends of chromosomes, which harbor the highest gene concentrations in the human genome, [3] are difficult to identify on routine chromosome analysis while they represent an important genetic cause of idiopathic ID. Therefore, testing for such rearrangements has turned out to be an important clinical evaluation step in the etiological diagnosis of unexplained ID cases. [4] In several studies, subtelomeric rearrangements were found to be associated with moderate to severe phenotypic abnormalities and turned out to be a significant cause of ID, with an estimated prevalence of 5-9% of cases in various populations. [5],[6],[7] To date, however, there are no data about the prevalence of subtelomeric rearrangements in Indonesia.
In a large number of Indonesian ID patients, the cause of ID could be established by conventional karyotyping or molecular testing for fragile X syndrome (FXS), but the majority of cases still remained unexplained. [8] Therefore, this study aimed at determining the prevalence of subtelomeric rearrangements and the clinical features in these ID individuals in Indonesia.
| Materials and Methods | | |
This research is an extension of previously reported studies on the identification of genetic causes of ID in Indonesia, where chromosomal aberrations and FXS were investigated in a large cohort of 527 Indonesian ID individuals from several special schools and institutions in Java Island, Indonesia. These previous studies revealed chromosomal abnormalities in 82 individuals and FXS in 9 individuals. [8],[9] In the present study, molecular testing of subtelomeric deletions and duplications was performed in the 436 as yet unresolved patients (278 males and 158 females). Informed consent was obtained from the parents or legal representatives, and the study has been approved by the Ethical Board of our institute. All subjects underwent a standardized clinical examination including physical measurements and dysmorphological assessment.
The DNA was isolated from peripheral blood using the salting out extraction procedure as described elsewhere. [10] Multiplex ligation dependent probe amplification (MLPA) analysis was performed as described previously. [11] Two probe-kits for subtelomeric chromosomal imbalances were used in these experiments: SALSA P070 and SALSA P036D MRC-Holland, Amsterdam, The Netherlands ( http://www.mrc-holland.com ). Each subtelomeric rearrangement was identified by at least one additional MLPA analysis using the SALSA P070 as the first level screening. Afterwards, SALSA P036D was utilized for confirmation of the aberration detected with the P070 kit. Rearrangements in specific regions were verified with SALSA kit P028, P023B, P340A or P096. The detail of regions detected by each kit is available at http://www.mlpa.com. Amplification products were identified and quantified by capillary electrophoresis on an ABI 3730 genetic analyzer (Applied Biosystems, Foster City, CA, USA) using GeneMapper Software version 3.7 (Apache Software; http://www.apache.org), manufacturer (University of Tokyo). Statistical analyses were carried out using Microsoft® Excel spreadsheets as described before. [12] The results were considered abnormal when the relative peak height ratio was below 0.70 or above 1.30.
When a deletion or duplication was detected in both MLPA kits, parents were tested for de novo occurrence. When parental DNA was not available, additional methodologies were performed for confirmation of the presence of the detected deletion or duplication. Fluorescence in situ hybridization (FISH) analysis was performed using commercially available probes (Vysis, Inc., Downers Grove, IL, USA) according to the manufacturer's recommendations. Single Nucleotide Polymorphism (SNP) array analyses were performed using the Affymetrix NspI 250K SNP array platform ( www.Affymetrix.com ). Copy number estimates were determined using the Copy Number Analyzer for Affymetrix Genechip Mapping (CNAG) software package version 2 ( http://www.genome.umin.jp/ ), manufacturer (University of Tokyo). [13]
The clinical data of all patients were reviewed and compared to other cases with a comparable aberration, described in the literature.
| Results | | |
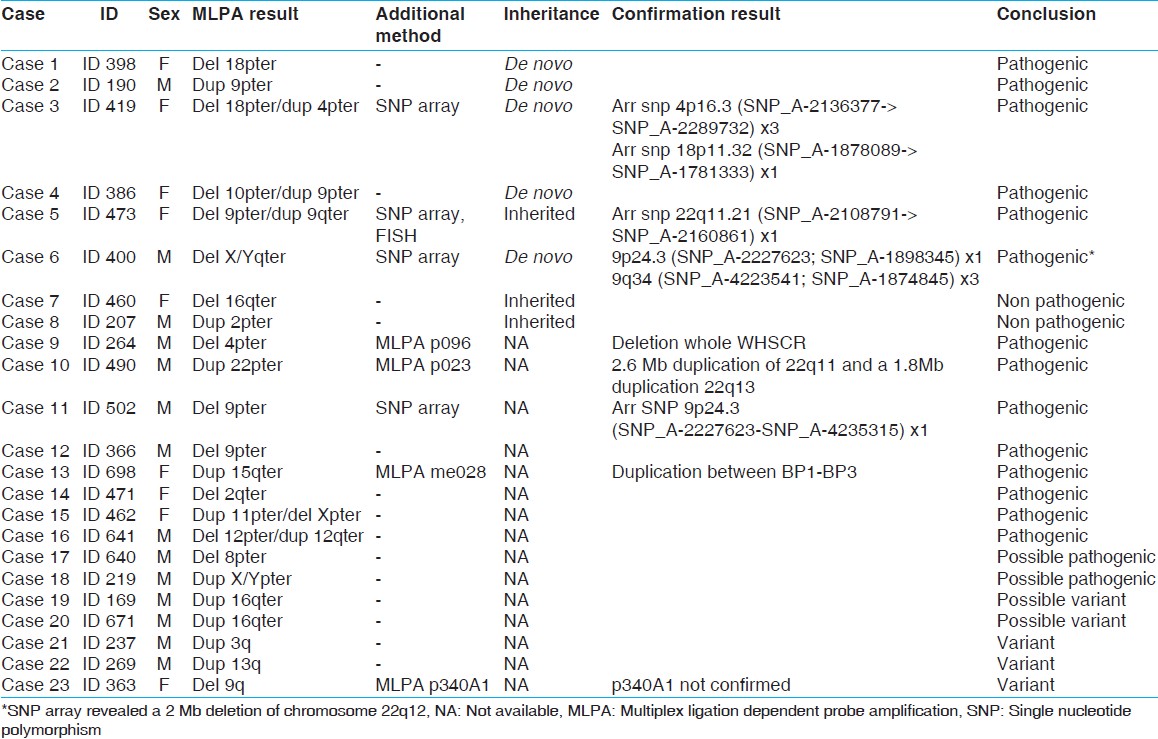
The initial screening with the SALSA P070 probe kit showed a subtelomeric deletion and/or duplication in 23 of the 436 ID individuals [Table 1]. In 20 of these samples, the presence of the aberration could be confirmed by the SALSA P036D kit [cases 1-20, [Table 1], while in the three remaining cases, this was not possible and they were considered to be either artefacts or non-causative variants. Parental testing was possible in eight of the 20 cases (cases 1-8) and revealed a de novo occurrence of the subtelomeric imbalances in five cases (cases 1-4 and 6). These five aberrations were, therefore, considered being pathogenic [Table 1] and [Table 2]. The phenotypes of cases 1 and 2 share many similarities with known cases with monosomy 18pter [14] or a subtelomeric duplication of 9p24, [15] respectively. In cases 3, 4, and 5, a subtelomeric deletion appeared to be coexistent with a subtelomeric duplication, which implicates the presence of a cryptic unbalanced translocation. The phenotype of case 3 was comparable to reported cases with either a 4p duplication [14] or a 18p deletion. [16] Therefore, either dup 4pter or deletion 18pter (or both) could be contributing to the phenotype in the case 3. In case 4, the phenotype is most likely due to the deletion of 10pter, because of the phenotypic overlap with previously reported cases with a deletion of 10p15. [17] The dysmorphic features of this case do not match the clinical description of another individual with a duplication of 9pter [15] In case 5, the 9pter/qter deletion/duplication is considered to be causative. The clinical details have been reported elsewhere. [18] In case 6, a de novo deletion of the X/Yqter pseudo autosomal region 2 was detected, which was previously also described in phenotypically normal individuals (DGV; http://www.tcag.ca/). Therefore, we performed an additional SNP array analysis to enable a fairly precise determination of the size of the deletion and link it to the severity of the clinical features. To our surprise, this revealed a 2 Mb deletion on chromosome 22q11 that has previously been described in patients with a similar phenotype. [19] We therefore, conclude that the phenotype of case 6 is not due to the de novo X/Yqter deletion, but due to the 22q11 deletion.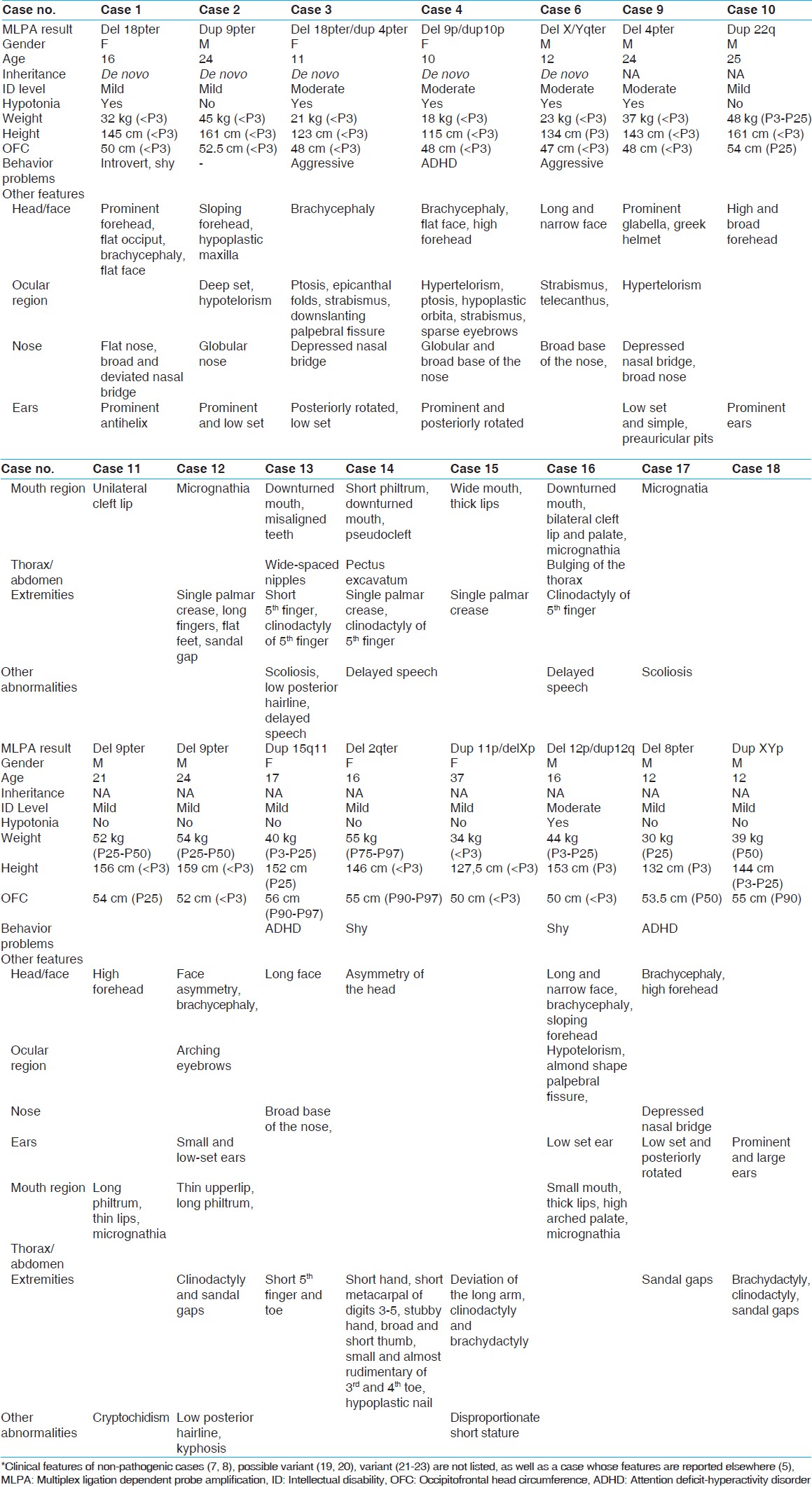 | Table 2: Clinical features of individuals with subtelomeric rearrangements*
Click here to view |
In two cases (case 7 and 8), parental testing showed that the aberration was inherited from an unaffected parent. These aberrations were therefore, considered being familial variants that do not contribute to the phenotype in case 7 and 8. When parental samples were not available, we tried to confirm the presence of the detected subtelomeric aberration using additional methodologies [Table 1]. The deletion of the entire Wolf-Hirschhorn Critical Region (WHSCR) in 4pter in case 9 was confirmed by MLPA with the SALSA P096 probe kit. Furthermore, the clinical features of this patient were consistent with Wolf-Hirschhorn Syndrome. In case 10, two duplicated regions of chromosome 22 were identified, one in the 22q11.2 region (next to the centromere) and the other in the 22q13.3 region (telomere end of q arm of chromosome 22). The SALSA P023B probe kit was used to confirm these duplicated regions. Two duplications in one arm of the chromosome suggested a complex recombination. However, such recombination could not be identified in the routine analysis and warranted further characterization. Confirmatory analysis using SNP array showed that both duplications actually consisted of 2.6 Mb in 22q11.2 and 1.8 Mb in 22q13.33. Since microduplications of both 22q11.2 and 22q13.3 have been associated with highly variable phenotypic features, [20],[21] we suggest that in case 10, both duplications contribute to the phenotype.
In cases 11 and 12, a subtelomeric deletion of 9pter was identified. Case 11 showed a milder phenotype than previously reported cases. [22],[23] In order to see, if the deletion in case 11 was smaller than the deletions reported before, SNP array analysis was performed. The deletion appeared to be 11.8 Mb in size, and it does not exceed the critical region of 9p syndrome; [22],[23] this, therefore, explained the mild phenotype. In case 12, however, the patient showed some similarities to the reported cases. [22] Therefore, it is suggested that 9pter deletion in case 12 is causative, and array analysis to determine the actual size of the deletion was unnecessary.
In case 13, a microduplication of 15q11 was identified. MLPA analysis of probes in the 15q11.2-15q15.1 region (MRC Holland kit P028) showed a duplication of the probes between breakpoint 1 and breakpoint 3 (including TUBGCP5 and APBA2). The methylation specific analysis indicated that the interstitial duplication was of maternal origin. It is suggested, that the duplication explains mild ID and minor dysmorphic features in case 13, since duplications of 15q11 are associated with a highly variable phenotype. [24]
In cases 14-20, parental testing and additional testing were not performed. This was due to the fact that in some cases, materials for additional testing were unavailable. Another reason was that the clinical features of the patient showed some similarities to the reported cases; therefore, additional testing was considered unnecessary. In case 14, a subtelomeric deletion of 2qter was identified. This patient showed shortening of the metacarpal bones which occurs in the majority of 2q37 patients. [25] It is suggested, therefore that the deletion in this case is causative. In case 15, a subtelomeric deletion of Xpter and a subtelomeric duplication of 11pter were identified. The presence of both a deletion and duplication suggests an unbalanced translocation. The Xpter probes in the P070 and P036D MLPA kits are both located within the SHOX gene. Deletions of this gene are associated with Leri-Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). [26],[27] A duplication of 11pter has been reported in a case of Silver- Russell syndrome More Details. [28] Therefore, both the dup 11pter and the deletion Xpter could be contributing to the short stature of this individual's phenotype.
In case 16, the subtelomeric deletion of 12pter accompanied by a subtelomeric duplication of 12qter suggests that the recombinant chromosome resulted from a pericentric inversion in one of the parents. Unfortunately, his parents were unavailable to test. Lagier-Tourenne et al. (2004) reported two cases of microscopically visible recombinant chromosome 12 and reviewed all previously reported cases as well as cases with pure cytogenetic deletion of 12pter and duplication of 12qter. [29] Compared to these previously reported cases with larger abnormalities, our patient showed a milder phenotype such as minor facial dysmorphisms and mild ID [Table 2]. It is suggested that the terminal duplication/deletion of chromosome 12 in this patient was smaller than previously reported and could contribute to the phenotype.
In case 17, a subtelomeric deletion of 8pter was identified. He showed mild ID with minor facial dysmorphisms. A subtelomeric 8pter deletion is rare, and only few cases have been reported. [4] Wu et al. (2010) reported a patient with a very small deletion in terminal 8pter with ID, microcephaly and minor facial dysmorphisms. We therefore conclude that the clinical features of case 17 are most likely due to the deletion of 8pter.
In case 18, a duplication of the probes in the SHOX gene in the pseudo-autosomal region 1 (PAR1) Xpter/Ypter was identified. SHOX duplications limited to PAR1 appear to be rare, and the associated phenotype is highly variable. [30],[31] SHOX gene defects, either a deletion or duplication, were associated with LWD and ISS. It has to be noted; however, that the effect of a duplication is ambiguous. [27] Consequently, the clinical features associated with such duplication were likely to be under-ascertained. [30] We are uncertain, therefore, whether the duplication of this gene contributed to the clinical phenotype or not.
In cases 19 and 20, a subtelomeric duplication of 16qter was identified. A 16qter submicroscopic microduplication is rarely reported. Ravnan et al. (2006) reported five cases with a duplication 16qter in which the duplicated signal was adjacent to the 18p subtelomere probe signal. In two cases, the recombination appeared to be inherited from unaffected parents, and these were considered to be variants. Therefore, in the other three cases the recombination was also regarded to be a variant although parental samples were not available. [32] It cannot be ruled out; however, that the duplication in cases 19 and 20 are contributing to their phenotype since 16qter is a gene-rich region. More than ten genes are present in the ~500 kb proximal to 16qter. Some of these (NULP1, TUBB3, and AFG3L1) are expressed in the brain, [33] and it is possible that overexpression of these genes contributes to ID.
| Discussion | | |
This is the first study identifying subtelomeric chromosomal aberrations in Indonesian ID individuals. Overall, subtelomeric copy number rearrangements were established in 20 samples, explaining the phenotype of 16 cases. Therefore, a detection rate of 3.7% (16/436) was obtained, of which 31% (5/16) was found to have a complex rearrangement/unbalanced translocation, 44% (7/16) had a simple deletion and 25% (4/16) had a simple duplication. In addition, the subtelomeric rearrangements contributed as genetic cause of ID in 3% (16/527) of cases in the whole cohort. The deletions, including the complex rearrangements, involved nine different subtelomeric regions (2q, 4p, 8p, 9p, 10p, 12p, 18p, X/Yp, X/Yq); and duplications, including complex rearrangements, involved eight subtelomeric regions (4p, 9p, 9q, 11p, 12q, 15q11, 22p, X/Yp).
The detection rate of chromosomal subtelomeric rearrangements in this study is 3.7% (16/436) which is well within the range of 2.5% previously reported by Ravnan et al., [32] and 4.4% as reported by van Karnebeek et al. [2] Five individuals (5/16; 31%) are suggested to have an unbalanced translocation that was not detected by routine cytogenetic analysis. In three of these, the translocation was shown to be de novo (case 3, case 4, and case 6), whereas in the two others, parental samples were unavailable. The prevalence of these cryptic imbalances in our ID population is in the range of a previous study conducted by Wu et al. (2010), who reported such rearrangements in 21.7% (5/23) and the study of Jehee et al. (2011) which reported such rearrangements in 42.1% (8/19). [6],[7] The considerably high rate of unbalanced translocations observed in this study might be explained by the fact that we did not use high-resolution banding, which could have detected most "cryptic" subtelomeric anomalies. [8]
Although the MLPA method is capable of revealing subtelomeric rearrangements, the clinical significance of each rearrangement should be interpreted carefully, particularly for cases in which the clinical features are different from previously reported cases. In case 6, for example, the de novo subtelomeric X/Yqter deletion could not explain the clinical features when compared to the previously reported cases. [32],[34] Furthermore, in the DGV it is reported that rearrangements in this region can be detected in phenotypically normal individuals as well. Subsequent SNP array in this patient identified another abnormality, which explained his clinical phenotype.
To conclude, this is the first large-scale study of the detection of submicroscopic subtelomeric aberrations in Indonesian patients with ID. This study shows that subtelomeric rearrangements are an important cause of ID in Indonesia, and its prevalence does not differ from previously reported studies in the Western world. Since diagnostic facilities for this kind of abnormalities are not yet available in Indonesia, the implementation of this technique in a routine diagnostic setting will help to establish a genetic diagnosis in individuals with ID, and will improve the possibilities for genetic counseling to the families involved. To establish an adequate diagnosis is of crucial importance for patients and their families. Therefore, diagnostic facilities for genetic diseases need to get a higher priority in Indonesia, similar to those for common infectious diseases and nutritional problems.
| Acknowledgment | | |
We thank all participants and their families for their contribution. We also thank laboratory staff at Department of Human Genetics, RUNMC, The Netherlands and CEBIOR, FMDU, Semarang Indonesia, for their expert technical assistance, in particular Marga Schepens and Alfi Afadiyanti.
| References | | |
| 1. | Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet 2010;11:161-87. 
|
| 2. | van Karnebeek CD, Jansweijer MC, Leenders AG, Offringa M, Hennekam RC. Diagnostic investigations in individuals with mental retardation: A systematic literature review of their usefulness. Eur J Hum Genet 2005;13:6-25.
|
| 3. | Rudd MK. Structural variation in subtelomeres. Methods Mol Biol 2012;838:137-49.
|
| 4. | De Vries BB, Winter R, Schinzel A, van Ravenswaaij-Arts C. Telomeres: A diagnosis at the end of the chromosomes. J Med Genet 2003;40:385-98.
|
| 5. | Christofolini DM, de Paula Ramos MA, Kulikowski LD, da Silva Bellucco FT, Belangero SI, Brunoni D, et al. Subtelomeric rearrangements and copy number variations in people with intellectual disabilities. J Intellect Disabil Res 2010;54:938-42.
|
| 6. | Jehee FS, Takamori JT, Medeiros PF, Pordeus AC, Latini FR, Bertola DR, et al. Using a combination of MLPA kits to detect chromosomal imbalances in patients with multiple congenital anomalies and mental retardation is a valuable choice for developing countries. Eur J Med Genet 2011;54:e425-32.
|
| 7. | Wu Y, Ji T, Wang J, Xiao J, Wang H, Li J, et al. Submicroscopic subtelomeric aberrations in Chinese patients with unexplained developmental delay/mental retardation. BMC Med Genet 2010;11:72.
|
| 8. | Mundhofir FE, Winarni TI, van Bon BW, Aminah S, Nillesen WM, Merkx G, et al. A cytogenetic study in a large population of intellectually disabled Indonesians. Genet Test Mol Biomarkers 2012;16:412-7.
|
| 9. | Mundhofir FE, Winarni TI, Nillesen WM, van Bon BW, Schepens M, Ruiterkamp-Versteeg M, et al. Prevalence of fragile X syndrome in males and females in Indonesia. World J Med Genet 2012;2:15-22.
|
| 10. | Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988;16:1215.
|
| 11. | Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002;30:e57.
|
| 12. | Koolen DA, Nillesen WM, Versteeg MH, Merkx GF, Knoers NV, Kets M, et al. Screening for subtelomeric rearrangements in 210 patients with unexplained mental retardation using multiplex ligation dependent probe amplification (MLPA). J Med Genet 2004;41:892-9.
|
| 13. | Nannya Y, Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, et al. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res 2005;65:6071-9.
|
| 14. | Brenk CH, Prott EC, Trost D, Hoischen A, Walldorf C, Radlwimmer B, et al. Towards mapping phenotypical traits in 18p-syndrome by array-based comparative genomic hybridisation and fluorescent in situ hybridisation. Eur J Hum Genet 2007;15:35-44.
|
| 15. | Ruiter EM, Koolen DA, Kleefstra T, Nillesen WM, Pfundt R, de Leeuw N, et al. Pure subtelomeric microduplications as a cause of mental retardation. Clin Genet 2007;72:362-8.
|
| 16. | Cyr AB, Nimmakayalu M, Longmuir SQ, Patil SR, Keppler-Noreuil KM, Shchelochkov OA. A novel 4p16.3 microduplication distal to WHSC1 and WHSC2 characterized by oligonucleotide array with new phenotypic features. Am J Med Genet A 2011;155A: 2224-8.
|
| 17. | Lindstrand A, Malmgren H, Verri A, Benetti E, Eriksson M, Nordgren A, et al. Molecular and clinical characterization of patients with overlapping 10p deletions. Am J Med Genet A 2010;152A: 1233-43.
|
| 18. | Mundhofir FE, Smeets D, Nillesen WM, Winarni TI, Yntema HG, Leeuw N, et al. Monosomy 9pter and trisomy 9q34.11qter in two sisters due to a maternal pericentric inversion. Gene 2012;511(2):451-4.
|
| 19. | Repetto GM, Guzmán ML, Puga A, Calderón JF, Astete CP, Aracena M, et al. Clinical features of chromosome 22q11.2 microdeletion syndrome in 208 Chilean patients. Clin Genet 2009;76:465-70.
|
| 20. | Feenstra I, Koolen DA, Van der Pas J, Hamel BC, Mieloo H, Smeets DF, et al. Cryptic duplication of the distal segment of 22q due to a translocation (21;22): Three case reports and a review of the literature. Eur J Med Genet 2006;49:384-95.
|
| 21. | de La Rochebrochard C, Joly-Hélas G, Goldenberg A, Durand I, Laquerrière A, Ickowicz V, et al. The intrafamilial variability of the 22q11.2 microduplication encompasses a spectrum from minor cognitive deficits to severe congenital anomalies. Am J Med Genet A 2006;140:1608-13.
|
| 22. | Hauge X, Raca G, Cooper S, May K, Spiro R, Adam M, et al. Detailed characterization of, and clinical correlations in, 10 patients with distal deletions of chromosome 9p. Genet Med 2008;10:599-611.
|
| 23. | Swinkels ME, Simons A, Smeets DF, Vissers LE, Veltman JA, Pfundt R, et al. Clinical and cytogenetic characterization of 13 Dutch patients with deletion 9p syndrome: Delineation of the critical region for a consensus phenotype. Am J Med Genet A 2008;146A:1430-8.
|
| 24. | Bolton PF, Dennis NR, Browne CE, Thomas NS, Veltman MW, Thompson RJ, et al. The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am J Med Genet 2001;105:675-85.
|
| 25. | Felder B, Radlwimmer B, Benner A, Mincheva A, Tödt G, Beyer KS, et al. FARP2, HDLBP and PASK are downregulated in a patient with autism and 2q37.3 deletion syndrome. Am J Med Genet A 2009;149A: 952-9.
|
| 26. | Barroso E, Benito-Sanz S, Belinchón A, Yuste-Checa P, Gracia R, Aragones A, et al. Identification of the first de novo PAR1 deletion downstream of SHOX in an individual diagnosed with Léri-Weill dyschondrosteosis (LWD). Eur J Med Genet 2010;53:204-7.
|
| 27. | Hirschfeldova K, Solc R, Baxova A, Zapletalova J, Kebrdlova V, Gaillyova R, et al. SHOX gene defects and selected dysmorphic signs in patients of idiopathic short stature and Léri-Weill dyschondrosteosis. Gene 2012;491:123-7.
|
| 28. | Eggermann T, Spengler S, Bachmann N, Baudis M, Mau-Holzmann UA, Singer S, et al. Chromosome 11p15 duplication in Silver-Russell syndrome due to a maternally inherited translocation t (11;15). Am J Med Genet A 2010;152A:1484-7.
|
| 29. | Lagier-Tourenne C, Ginglinger E, Alembik Y, De Saint Martin A, Peter MO, Dulucq P, Jonveaux P, Jeandidier E. Two cousins with partial trisomy 12q and monosomy 12p recombinants of a familial pericentric inversion of the chromosome 12. Am J Med Genet. 2004;125A: 77-85.
|
| 30. | Hirschfeldova K, Baxova A, Kebrdlova V, Solc R, Mihalova R, Lnenicka P, et al. Cryptic chromosomal rearrangements in children with idiopathic mental retardation in the Czech population. Genet Test Mol Biomarkers 2011;15:607-11.
|
| 31. | Thomas NS, Harvey JF, Bunyan DJ, Rankin J, Grigelioniene G, Bruno DL, et al. Clinical and molecular characterization of duplications encompassing the human SHOX gene reveal a variable effect on stature. Am J Med Genet A 2009;149A: 1407-14.
|
| 32. | Ravnan JB, Tepperberg JH, Papenhausen P, Lamb AN, Hedrick J, Eash D, et al. Subtelomere FISH analysis of 11 688 cases: An evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J Med Genet 2006;43:478-89.
|
| 33. | Zou YS, Van Dyke DL, Ellison JW. Microarray comparative genomic hybridization and FISH studies of an unbalanced cryptic telomeric 2p deletion/16q duplication in a patient with mental retardation and behavioral problems. Am J Med Genet A 2007;143:746-51.
|
| 34. | Parvari R, Mumm S, Galil A, Manor E, Bar-David Y, Carmi R. Deletion of 8.5 Mb, including the FMR1 gene, in a male with the fragile X syndrome phenotype and overgrowth. Am J Med Genet 1999;83:302-7.
|
[Table 1], [Table 2]
|