|
 
 |
|
ORIGINAL ARTICLE |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 2 | Page : 207-212 |
| |
Unique pattern of mutations in β-thalassemia patients in Western Uttar Pradesh
Ajay F Christopher1, Anita Kumari2, Sunali Chaudhary1, Sandhya Hora1, Ziledar Ali1, Satish C Agrawal2
1 Central Research Laboratory, Department of Biochemistry, S.R.M.S. Institute of Medical Sciences, Bareilly, Uttar Pradesh, India
2 Department of Pediatrics, S.R.M.S. Institute of Medical Sciences, Bareilly, Uttar Pradesh, India
| Date of Web Publication | 5-Aug-2013 |
Correspondence Address:
Anita Kumari
Department of Pediatrics, S.R.M.S. Institute of Medical Sciences, Bareilly - 243 202, Uttar Pradesh
India
 Source of Support: S.R.M.S. Institute of Medical Sciences, Bareilly, Uttar Pradesh, India, Conflict of Interest: None
DOI: 10.4103/0971-6866.116119

 Abstract Abstract | | |
Context: β-thalassemia is one of the most common heterogeneous inherited single gene disorders. The disease results from one or more of 380 different mutations in the β-globin gene. Uttar Pradesh (U.P.) is the most populous state of India, comprising various ethnic groups and Bareilly is one of the largest cities situated in Western U.P.
Aims: To examine the prevalence of five common β-thalassemian mutations: Intervening Sequence IVS 1-5 (c. 92 + 5 G > C), codon 8/9 (c. 27_28insG), codon 41/42 (c. 124_127delTTCT), IVS 1-1 (c. 92 + 1 G > T) and codon 26 G-A (c. 79G > A) in Western U.P.
Settings and Design: Patients attending camps organized by the Thalassemia Society, Bareilly were selected for the study.
Materials and Methods: A total of 48 blood samples were collected from the patients of transfusion dependent β-thalassemia from July 2011 to May 2012. All the samples were analyzed for five common mutations by using the Amplification Refractory Mutation System (ARMS)-hot start-polymerase chain reaction (PCR) technique.
Results: Among the five common mutations prevalent in India, we were able to detect all except codon 26 G-A (c. 79G > A), which is prevalent in northeast India. These four mutations accounted for 58% of the total number of our patients. The IVS 1-5 (G-C) was found to be the most common mutation with a frequency of 46% and the 2 nd most common mutation was Fr8/9 (+G) with a frequency of 21%. The frequency of other mutations was IVS1-1 (12%) and Cd 41/42 (4%).
Conclusion: This study provides evidence that the pattern of mutations in Western U.P. is different from the rest of India and even from the neighboring states (Delhi and Punjab). To the best of our knowledge, mutation Fr8/9, the 2 nd most common mutation in our study has never been reported to be so common from anywhere in India. Some mutations, which are prevalent in other regions are absent in our region (mutation for ε-globin). Hence, these findings can be called unique to Western U.P.
Keywords: Hot start polymerase chain reaction, mutation, prenatal diagnosis, β-thalassemia
How to cite this article:
Christopher AF, Kumari A, Chaudhary S, Hora S, Ali Z, Agrawal SC. Unique pattern of mutations in β-thalassemia patients in Western Uttar Pradesh. Indian J Hum Genet 2013;19:207-12 |
How to cite this URL:
Christopher AF, Kumari A, Chaudhary S, Hora S, Ali Z, Agrawal SC. Unique pattern of mutations in β-thalassemia patients in Western Uttar Pradesh. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:207-12. Available from: http://www.ijhg.com/text.asp?2013/19/2/207/116119 |
| Introduction | |  |
β-thalassemia is the most common autosomal single gene disorder across the globe. [1],[2] This disorder is caused by a deficient (β+ thalassemia) or absent (β° thalassemia) synthesis of one or more of the globin chains. Until date, more than 380 different types of mutations causing this disease have been identified in Online Mendalian Inheritance in Man (OMIM) 2005. The endemic area of the disease is the "thalassemia belt," which includes the Mediterranean region, part of the Middle-East, the Indian subcontinent and the South-East Asia. [3],[4] Immigration plays a major role in both the distribution and the extent of mutations across the globe. [5] There are approximately 45 million carriers with a carrier rate of approximately 1:20, of β-thalassemia in South Asian countries which include India, Sri Lanka, and Pakistan. [6] Treatment of individuals with β-thalassemia major, which entails regular blood transfusions and expensive iron chelation regimen, is still not satisfactory. Thus, the disease causes significant morbidity and mortality in affected individuals. [7] β-thalassemia is a major public-health problem and a burden on the society. The cost for blood transfusions alone for the disease has been projected at ~3,200 USD (equivalent to approximately 1,76,000 Indian rupees) per child, while the lifetime health-care for a person born with the disease has been projected at 284,154 USD (1,56,28,000 Indian rupees). [8]
Though, β-thalassemia is observed in most global populations, each population or sub population has its own unique spectrum of mutations. [9] In India, the estimated number of β-thalassemia heterozygous patients is about 29.7 million with a rate of about 7000 homozygous children born with this disease per year. [10] The frequency of the heterozygous carriers reaches as high as 40% in some tribal groups, whereas, the incidence of the disease is around 3-4% in the general population. [11],[12] About 92% of β-thalassemia allele in India is accounted by common mutations such as IVS 1-5 (c. 92 + 5 G > C), IVS 1-1 (c. 92 + 1 G > T), 619 bp deletion, codon 8/9 (c. 27_28insG), and codon 41/42 (c.124_127delTTCT). [13] The most common mutation across India is IVS 1-5 (c. 92 + 5 G > C) as also found in Pakistan and Sri Lanka. These mutations are not uniformly distributed across India but have geographic specificity, and each ethnic group has some common mutations and a variable number of rare ones. Regional differences in prevalence across northern, eastern and southern parts of India have also been demonstrated. In the northern states of India, the mutation and allele frequencies are quite similar to those in Pakistan where the two most common mutations are IVS 1-5 (43.5%) and codon 8/9 (38.5%). While in South India [14] codon 41/42 (c. 124_127delTTCT) is prevalent, in Eastern India mutation HBE (codon 26 G-A) (c. 79G > A) is frequently found. [15]
The β-globin chain is encoded by the hemoglobin β (HBB) gene. The β-globin locus harbors, in addition to the HBB gene, the ε-globin (HBE) gene, the two γ-globin (HBG) genes (HBG-G and HBG-A) and the δ-globin (HBD) gene. The most genes at this locus are expressed at specific time points in development. During embryonic development, the HBE gene is expressed along with the α-globin gene to form embryonic hemoglobin or HBE. 12 weeks post-conception, the fetus primarily uses fetal hemoglobin (Hb-F), which is composed of two HBG and two α-globin chains. Around the time of birth, HBG production decreases while β-globin synthesis increases so that most individuals have only trace amounts of Hb-F detectable 7-8 months after birth. The combination of two α-globin chains and two β-globin chains makes a normal adult Hb, which becomes the predominant form within 18-24 weeks after birth. Less than 3% of adult hemoglobin is Hb type A2, which is composed of two HBD chains and two α-globin chains-globin chains. [16],[17] Therefore, any test other that nucleic acid based investigation cannot be used for prenatal diagnosis.
β-thalassemias are heterogenous at the molecular level. [18] Majority of mutations are single nucleotide substitutions, deletions or insertions of olegonucleotides leading to frameshift; rarely it results from gross gene deletion. Point mutations affecting the β-globin expression belong to three different categories: Mutations leading to defective β-gene transcription (promotor and 5' UTR mutations); mutations affecting messenger RNA (mRNA) processing (splice-junction and consensus sequence mutations, polyadenylation, and other 3' UTR mutations); and mutations resulting in abnormal mRNA translation (nonsense, frameshift, and initiation codon mutations). β°-thalassemias are characterized by the complete absence of β-chain production, which results from deletion, initiation codon, nonsense, frameshift, and splicing mutations, especially, at the splice-site junction. On the other hand, β+ -thalassemias are characterized by reduced production of the β-chains, which are produced by mutations in the promoter area, the polyadenylation signal, and the 5'or 3' UTR or by splicing abnormalities.
The population of U.P. is over 200 million; it is a multi-ethnic population but marriages most often take place within the same ethnic groups. As β-thalassemia is inherited in an autosomal recessive manner, couples who are both heterozygous for β-thalassemia gene carry a 25% risk of having a child with β-thalassemia major (homozygous for β-thalassemia gene). Information provided on the distribution and the frequency of β-thalassemia alleles is useful to establish a program for carrier screening, genetic counseling, prenatal diagnosis, and for physicians to establish specific therapeutic approaches for patients suffering from the disease. Due to lack of information regarding the type of β-thalassemia mutations encountered in Western U.P., an effort was made to characterize the mutations for β-thalassemia by ARMS-hot start PCR.
| Materials and Methods | | |
Specimens
The sample consisted of 48 unrelated individuals (32 males and 16 females) with age ranging between 2 years and 16 years. The patients included in this study were all transfusion dependent. Age, sex, history, and consanguinity between the parents were recorded by reviewing the patients' files after obtaining permission from their parents. The age at presentation of the disease was as early as 6 months. Most of these patients had received transfusion 14-16 times/year, but only a few of them had received transfusion at irregular intervals due to economic and other constraints. Samples of venous blood (2 ml) from each patient was collected in tubes containing ethylene diamine tetraacetic acid (EDTA) (Volex, Italy) prior to blood transfusion during thalassemia camps organized by the Thalassemia Society, Bareilly between July 2011 and May 2012.
DNA extraction and characterization of mutations by ARMS-PCR
DNA was isolated from whole blood using HiPurA blood genomic DNA midiprep purification Spin Kit (Himedia, Mumbai). RNA was digested with 200 μl of RNase A solution (20 mg/ml, Himedia, Mumbai). The isolated DNA was evaluated by agarose gel electrophoresis after staining with ethidium bromide and exposure to ultraviolet (UV) light and stored at − 20° C until mutation analysis. All the samples were then screened for the five most common mutations previously reported in Indian population by ARMS-hot start PCR.
Hot start-PCR reactions were carried in two separate tubes for the each sample, one tube for the amplification by the normal ARMS primer and the 2 nd for the amplification by the mutant ARMS primer. However, amplification in both tubes was carried out using a common reverse primer. To monitor false-negative results, an internal PCR control, which amplified another region of the genome was included in the reaction. ARMS-PCR primers were synthesized by Bangalore genei, Bangalore. The sequences of allele-specific oligonucleotide primers (normal and mutant) are listed in [Table 1].
A total of 25 μl final PCR reaction volume was made in two separate tubes for each sample. The reaction mixture comprised 0.5 μg of the DNA template, 10 pmol of each of the four primers (two control primers, one mutant ARMS primer or one normal ARMS primer and its corresponding reverse primer for the reaction) in a solution of 10 mM Tris-HCl, 50 mM KCl and 1.2 mM MgCl 2 (Himedia, Mumbai). For hot start PCR, Taq DNA polymerase 2.5 units (Himedia, Mumbai) and 0.2 mM of each deoxyribonucleotide triphosphate (Bangalore genei, Bangalore) were added into each tube after the initial denaturation step (96° C for 1 min) on a thermal cycler (XP thermal cycler, Bioer, Japan). The reaction tubes were then subjected to thermal cycling regimen consisting of 25 cycles; denaturation at 94° C for 1 min, primer annealing at 65°C for 1 min and extension at 72°C for 1.5 min with the final extension at 72° C for 3 min.
The electrophoresis condition was as follows. Fifteen microlitres of the PCR products was removed and mixed with 3 μl of a loading buffer and then loaded on a 2% agarose gel and the gel was set at 100 volts for 1 h and then stained with ethidium bromide. After staining, the bands were visualized under UV light. Images of the gels were acquired on DigiDoc-It TM imaging system (UVP, USA) and molecular weight of the PCR products (test mutation) were determined by comparing them with the 100 base pair (bp) DNA marker using the Doc-It® LS image acquisition and analysis software (UVP, USA).
| Results | | |
The DNA extracted [Figure 1] from the whole blood samples of transfusion dependent β-thalassemia patients were subjected to ARMS-hot start PCR for detection of mutations. This technique is one of the most commonly used techniques for the diagnosis of the disease. The ARMS-hot start PCR results revealed the presence of 4 mutations [Table 2]. Internal control band of 861 bp was present in all ARMS-PCR reactions. Presence of an amplified product by the mutant ARMS primer on 2% agarose gel indicated the presence of the mutant allele. In the present study, we were able to identify the different β-thalassemia mutations in 28 patients (58%), while mutations were uncharacterized in 20 patients (42%). The reason for the uncharacterized mutations in these patients could be the presence of mutation other than the five selected mutations or that these patients harbored other hemaglobinopathies. Molecular analysis showed that IVS1-5(G-C) was the commonest β-thalassemic mutation (46%) followed by Fr8/9 (+G), which had a frequency of 21%. The frequency of other mutations was IVS1-1 (12%) and Cd 41/42 (4%). We were not able to find mutation for Cd26 (HBE), which is the 2 nd most widespread mutation in the north eastern states of India [Table 2].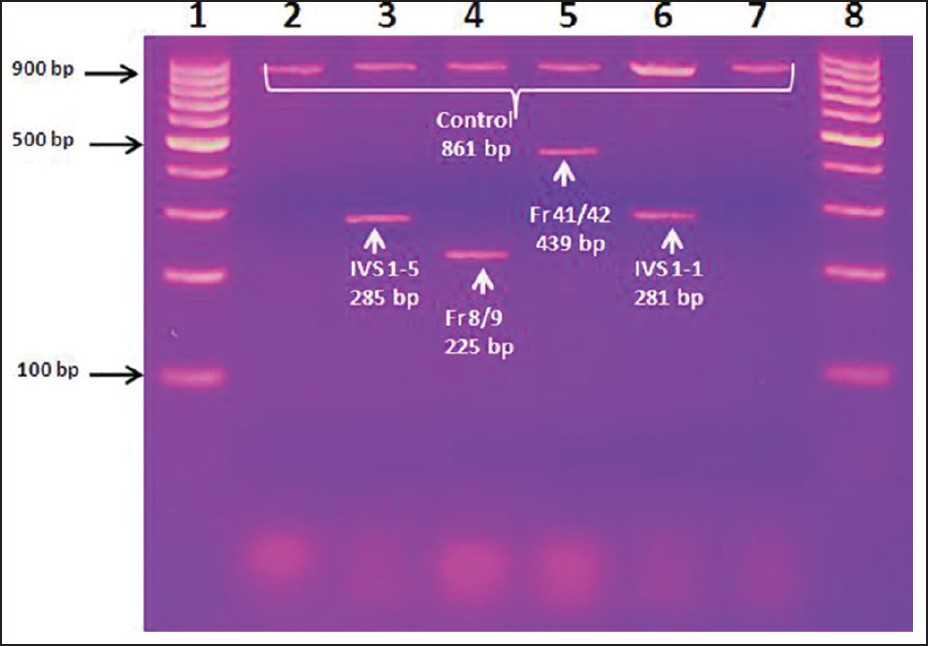 | Figure 1: ARMS - PCR gel for the diagnosis of β-thalassemia. DNAs extracted from the whole blood of thalassemic children were amplified by PCR using different primers and were run in agarose gel electrophoresis. the Ethidium Bromide stained gel was visualized on ultraviolet table; lane - 1: 100 bp DNA marker, lane - 3 IVS 1 - 5 mutation (285 bp) with control DNA (861 bp), lane - 4 Fr 8/9 mutation (225 bp), lane - 5 Fr 41/42 mutation (439 bp) and lane - 6 IVS 1 - 1 mutation (281 bp)
Click here to view |
| Discussion | | |
Of the 48 patients, we were able to detect four different type of mutations viz. IVS 1-5, CD 41/42, Fr 8/9 and IVS 1-1 in 28 (58%) patients. The most common mutation found in our study was IVS 1-5 (46%), which is in conformity with the previously reported studies as the most common mutation in the Indian population. [15],[19] Therefore, it can fairly be considered as the most common mutation in India.
Mutation Fr 8/9 was found to be the 2 nd most common mutation in our study with a frequency of 21%. This finding is in contrast with the previously reported data, which showed it as the fourth most common mutation in India. [10] Moreover, this mutation was not among the top three mutations in several other parts of the country, viz. Delhi, [20] Chandigarh, [21] Gujarat, [7] and Western India. [22] However, In Eastern India, this mutation ranked as the third most common, HBE being the 2 nd most common mutation, [15] while in South India, the same mutation has a very low frequency of occurrence. Hence, this mutation can be considered unique to Western U.P. High frequency of IVS 1-5 and Fr 8/9 mutations in our study suggests that these may be the oldest β-thalassemia mutations in Western U.P.
The 3 rd most common mutation in Western UP was IVS 1-1 (12%). This conforms with the data previously reported as the third most common mutation in India. [1] However, this mutation was not detected in Southern and Eastern India. [14],[15] The least common mutation detected in the present study was Cd 41/42 (4%); the same was reported from Delhi [20] but is in contrast with the data reported from Chandigarh. [21]
An important finding of the present study is that the distribution of Fr 8/9 mutation is high in comparison with the other regions of the country. To the best of our knowledge, this mutation as the 2 nd most prevalent mutation has never been reported previously from India. Hence, this mutation can be considered unique to Western U.P as HBE is unique to Eastern India.
β-thalassemia has been reported as probably the most common inherited hemoglobin disorder in the Indian subcontinent. [23] Since, no viable forms of treatment are available, the best course is prevention through prenatal diagnosis. Premarital screening in Indian population is still considered controversial, while the same is practiced successfully in Iran and Turkey. [24],[25] Although, the frequency of various β-thalassemia mutations in major cities in different states of India have already been reported, yet the common mutations in various rural areas of the country still need to be explored. Moreover, no attempt of molecular characterization of β-thalassemia has been carried out in Western U.P region. As each population or sub population has its own unique spectrum of mutations, in the present study, the prevalence of β-thalassemia mutations was investigated in-patients of this area. Through ARMS technique, we were able to analyze mutations in transfusion dependent β-thalassemia patients.
| Conclusion | | |
The people in U.P exhibit a genetic heterogeneity that is unparalleled in other regions of India. As only a few prevalent HBB mutations underlie the majority of patients with β-thalassemia in this area, a relatively cost-effective dedicated carrier screening method could be implemented. Furthermore, prenatal diagnosis and other preventive approaches may be the other important strategies in this regard.
| Acknowledgment | | |
We are deeply grateful to the management of SRMS IMS for the financial assistance. We are also thankful to the Thalassemia Society, Bareilly for providing the blood samples of the thalassemia patients.
| References | | |
| 1. | Quek L, Thein SL. Molecular therapies in beta-thalassaemia. Br J Haematol 2007;136:353-65. 
|
| 2. | Pan HF, Long GF, Li Q, Feng YN, Lei ZY, Wei HW, et al. Current status of thalassemia in minority populations in Guangxi, China. Clin Genet 2007;71:419-26.
|
| 3. | Cao A, Saba L, Galanello R, Rosatelli MC. Molecular diagnosis and carrier screening for beta thalassemia. JAMA 1997;278:1273-7.
|
| 4. | Weatherall DJ, Clegg JB. The β-Thalassemia Syndromes. 4 th ed. Oxford: Blackwell Scientific Publication; 2001. p. 635-87.
|
| 5. | Gupta A, Hattori Y, Agarwal S. Initiation codon mutation in an Asian Indian family. Am J Hematol 2002;71:134-6.
|
| 6. | Agarwal S, Gupta A, Gupta UR, Sarwai S, Phadke S, Agarwal SS. Prenatal diagnosis in beta-thalassemia: An Indian experience. Fetal Diagn Ther 2003;18:328-32.
|
| 7. | Nigam N, Munshi N, Patel M, Soni A. Distribution of β-thalassemia mutations and its correlation with α thalassemia in Gujarati families. Int J Hum Genet 2003;3:221-4.
|
| 8. | Ginsberg G, Tulchinsky T, Filon D, Goldfarb A, Abramov L, Rachmilevitz EA. Cost-benefit analysis of a national thalassaemia prevention programme in Israel. J Med Screen 1998;5:120-6.
|
| 9. | Old JM, Khan SN, Verma I, Fucharoen S, Kleanthous M, Ioannou P, et al. A multi-center study in order to further define the molecular basis of beta-thalassemia in Thailand, Pakistan, Sri Lanka, Mauritius, Syria, and India, and to develop a simple molecular diagnostic strategy by amplification refractory mutation system-polymerase chain reaction. Hemoglobin 2001;25:397-407.
|
| 10. | Verma IC, Saxena R, Thomas E, Jain PK. Regional distribution of beta-thalassemia mutations in India. Hum Genet 1997;100:109-13.
|
| 11. | Mohanty D, Mukherjee MB. Sickle cell disease in India. Curr Opin Hematol 2002;9:117-22.
|
| 12. | Mohanty D, Colah R, Gorakshakar A. New Delhi 2008. Jai Vigyan S and T mission project on community control of thalassemia syndromes-Awareness, screening, genetic counseling and prevention-A national multicentric task force study of Indian Council of Medical Research.
|
| 13. | Baig SM, Azhar A, Hassan H, Baig JM, Aslam M, Ud Din MA, et al. Prenatal diagnosis of beta-thalassemia in Southern Punjab, Pakistan. Prenat Diagn 2006;26:903-5.
|
| 14. | Bashyam MD, Bashyam L, Savithri GR, Gopikrishna M, Sangal V, Devi AR. Molecular genetic analyses of beta-thalassemia in South India reveals rare mutations in the beta-globin gene. J Hum Genet 2004;49:408-13.
|
| 15. | Gajra B, Chakrabarti S, Sengupta B, De M, Mukherjee S, Talukder G. Prevention of β-thalassemia major and E β-thalassemia by prenatal diagnosis in eastern India. Int J Hum Genet 2003;3:225-35.
|
| 16. | Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med 2005;353:1135-46.
|
| 17. | Cohen AR, Galanello R, Pennell DJ, Cunningham MJ, Vichinsky E. Thalassemia. Hematology Am Soc Hematol Educ Program 2004;1:14-34.
|
| 18. | Cao A, Galanello R. Beta-thalassemia. Genet Med 2010;12:61-76.
|
| 19. | Varawalla NY, Old JM, Sarkar R, Venkatesan R, Weatherall DJ. The spectrum of beta-thalassaemia mutations on the Indian subcontinent: The basis for prenatal diagnosis. Br J Haematol 1991;78:242-7.
|
| 20. | Madan N, Sharma S, Rusia U, Sen S, Sood SK. Beta-thalassaemia mutations in northern India (Delhi). Indian J Med Res 1998;107:134-41.
|
| 21. | Garewal G, Das R. Spectrum of β-thalassemia mutations in Punjabis. Int J Hum Genet 2003;3:217-9.
|
| 22. | Colah R, Gorakshakar A, Phanasgaonkar S, D'Souza E, Nadkarni A, Surve R, et al. Epidemiology of beta-thalassaemia in Western India: Mapping the frequencies and mutations in sub-regions of Maharashtra and Gujarat. Br J Haematol 2010;149:739-47.
|
| 23. | Sukumaran PK, Maser HR. The distribution of abnormal haemoglobins in the Indian population. In: Proceedings of the First Conference of the Indian Society of Human Genetics, Vol. 1. Human Population Genetics in India. Mumbai: Brient Longman; 1973. p. 91-111.
|
| 24. | Ghanei M, Adibi P, Movahedi M, Khami MA, Ghasemi RL, Azarm T, et al. Pre-marriage prevention of thalassaemia: Report of a 100,000 case experience in Isfahan. Public Health 1997;111:153-6.
|
| 25. | Keskin A, Türk T, Polat A, Koyuncu H, Saracoglu B. Premarital screening of beta-thalassemia trait in the province of Denizli, Turkey. Acta Haematol 2000;104:31-3.
|
[Figure 1]
[Table 1], [Table 2]
|