|
 
 |
|
ORIGINAL ARTICLE |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 4 | Page : 437-442 |
| |
Mutation analysis of mitogen activated protein kinase 1 gene in Indian cases of 46,XY disorder of sex development
Dhanjit Kumar Das, Subodh G Rahate, Bhakti P Mehta, Harshavardhan M Gawde, Parag M Tamhankar
Genetic Research Centre, National Institute for Research in Reproductive Health, Parel, Mumbai, Maharashtra, India
| Date of Web Publication | 4-Jan-2014 |
Correspondence Address:
Dhanjit Kumar Das
Genetic Research Centre, National Institute for Research in Reproductive Health, Jahangir Merwanji Street, Parel, Mumbai - 400 012, Maharashtra
India
 Source of Support: Government of India for providing financial grants for
the study, Conflict of Interest: None  | 2 |
DOI: 10.4103/0971-6866.124372

 Abstract Abstract | | |
Background: Determination of sex is the result of cascade of molecular events that cause undifferentiated bipotential gonad to develop as a testis or an ovary. A series of genes such as SRY, steroidogenic factor-1 (SF1), AR, SRD5 α, Desert hedgehog (DHH) etc., have been reported to have a significant role in development of sex in the fetus and secondary sexual characteristics at the time of puberty. Recently, mitogen activated protein kinase kinase kinase 1 (MAP3K1) gene was found to be associated with 46, XY disorders of sex development (DSD).
Aim: The present study is focused to identify mutations in MAP3K1 gene in the cohort of 10 Indian patients with 46,XY DSD including one family with two affected sisters. These patients were already screened for SRY, SF1 and DHH gene, but no mutation was observed in any of these genes.
Materials and Methods: The entire coding regions of MAP3K1 were amplified and sequenced using the gene specific primers.
Results and Discussions: Sequence analysis of MAP3K1 gene has revealed four variants including one missense, two silent and one deletion mutation. The missense mutation p.D806N was observed in four patients with hypospadias. Two patients showed the presence of silent mutation p.Q1028Q present in exon 14. Another silent mutation p.T428T was observed in a patient with gonadal dysgenesis. We have also observed one deletion mutation p. 942insT present in two patients. The pathogenicity of the missense mutation p.D806N was carried out using in-silico approach. Sequence homology analysis has revealed that the aspartate at 806 was found to be well-conserved across species, indicated the importance of this residue. The score for polyphen analysis of this mutation was found to be 0.999 indicating to be pathogenic mutation. Since, p.D806N mutation was found to be important residue; it might contribute to sexual development. We have reported the presence of mutations/polymorphism in MAP3K1 gene. All the mutations were found to be polymorphism upon comparing to single nucleotide polymorphism database. However, in-silico analysis of the missense mutation revealed to be a pathogenic mutation.
Keywords: Disorders of sex development, mitogen activated protein kinase kinase kinase 1, mutation
How to cite this article:
Das DK, Rahate SG, Mehta BP, Gawde HM, Tamhankar PM. Mutation analysis of mitogen activated protein kinase 1 gene in Indian cases of 46,XY disorder of sex development. Indian J Hum Genet 2013;19:437-42 |
How to cite this URL:
Das DK, Rahate SG, Mehta BP, Gawde HM, Tamhankar PM. Mutation analysis of mitogen activated protein kinase 1 gene in Indian cases of 46,XY disorder of sex development. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:437-42. Available from: http://www.ijhg.com/text.asp?2013/19/4/437/124372 |
| Introduction | |  |
Disorders of sex development (DSD) refer to congenital conditions in which development of chromosomal, gonadal or anatomical sex is atypical. [1] Among DSDs, some of them are expressed as congenital condition while in others it may be manifested during puberty due to hormonal rage. Clinical presentation observed in patients with DSD ranges from ambiguous genitalia to complete female phenotype. Patient with DSD may have dysgenetic wolffian structure such as dysgenetic testes, mild to severe penoscrotal hypospadias with or without chordee; whereas the dysgenetic mullerian structure can also be observed that ranges from absence to the presence of a fully developed uterus and Fallopian tube More Detailss.
Among chromosomal abnormalities, DSDs are determined by presence/absence or numerical abnormality of sex chromosome (X and Y). However some of the conditions are due to single gene disorder. SRY gene present in the Y chromosome is the major male sex determining gene. Although the female development was thought to be a default in the absence of SRY (or Y chromosome); however in recent studies, it has been discovered that the presence of DAX1 gene on X chromosome accounts for female phenotype in mice. [2] Gonadal sex determination takes place when the bipotential gonadal ridge turns into either testes or ovaries. The process starts around 4 th week and ends by 12 th week of pregnancy and is directed in a critically timed and gene dosage dependent manner. SRY gene stimulates the bipotential gonadal ridge to develop into testes. Testes produce two hormones testosterone and anti-mullerian hormone (AMH). Testosterone and its derivative dihydrotestosterone induce the formation of wolffian structures while AMH degenerates the mullerian duct; therefore, manifests male phenotype. In females (46, XX) who do not possess the SRY gene, the ovary forming pathway is activated by a different set of proteins. The fully developed ovary then produces estrogen which triggers the development of uterus, oviducts and cervix from the mullerian duct. Along with SRY gene, various autosomal and sex-linked genes such as SOX9, WT1, steroidogenic factor-1 (SF1), AR, DHH and MAP3K1 are also known to be involved in the process of sex development of the fetus.
Our present study is focused on identification of mutations in mitogen activated protein kinase kinase kinase 1 (MAP3K1) gene that belongs to MAPK family of genes, which is known to be involved in transmitting signals generated at the cell surface into the cytosol and nucleus. [3] MAPKs comprise a family of ubiquitous proline-directed, protein-serine/threonine kinases, which participate in signal transduction pathways that control intracellular events including acute responses to hormones and major developmental changes in organisms. [4] Recently, MAP3K1 was first associated with 46,XY DSD by Pearlman et al. [5] However, Warr et al. has demonstrated from their studies that MAP3K1 has insignificant role in testis determination of mice. [6] Taking into consideration that the MAP3K1 gene is required for development, therefore, MAP3K1 is one of the suspected genes that might cause downstream alterations in development of sex in humans. In this study, mutation analysis of MAP3K1 gene was carried out in 10 cases of DSD.
| Materials and Methods | | |
Patients
All patients screened in this study were sporadic. 10 patients with 46,XY DSD were selected for the study. Patients were classified according to gender identity disorder in children listed under Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition. The clinical presentations of all the patients were given in [Table 1].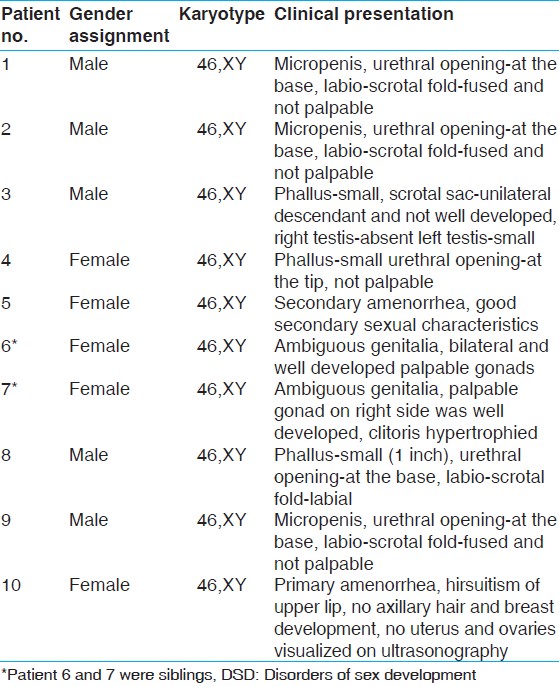 | Table 1: Clinical presentation of 46,XY DSD patients along with the gender assignment and karyotype
Click here to view |
Genomic deoxyribonucleic acid isolation and polymerase chain reaction amplification
Whole blood samples from patients and controls were collected in EDTA/K2 BD Vacutainer® tubes (Becton Dickinson and Company, NJ, USA). Genomic DNA was isolated from 2.0 ml of blood collected from the above patients using QIAmp DNA extraction kit (Qiagen, GmBH, Germany). PCR amplification was performed using Takara Hot Start PCR kit in 50 μl of ×10 Ex Taq™ buffer (contains 20 mM MgCl 2) , 10 mM deoxyribonucleotide triphosphate mixture (2.5 mM of each deoxyadenosine triphosphate, deoxycytidine triphosphate, deoxyguanosine triphosphate and deoxythymidine triphosphate), ×2.5 GC solution , 25 pmol primers, 250 ng template DNA and 1.5 units of Taq™ HS DNA polymerase. All the 21 exons were amplified separately using specific primers designed from the wild-type MAP3K1 sequence. PCR was cycled 35 times; each cycle consisted of denaturation for 1 min at 94°C, annealing for 45 min at 53-69°C and extension for 1 min at 72°C. After amplification, 3 μl of PCR product was subjected to electrophoresis on 2% agarose gel for 45 min at 100 V in tris-acetate-EDTA buffer and bands were stained with ethidium bromide (0.5 mg/ml).
Sequencing and analysis of sequence
The PCR products were gel purified using QIAquick Gel extraction kit (QIAGEN, GmBH, Germany) according to manufacturer's protocol. The gel purified PCR products were sequenced using gene specific primers on ABI PRISM 3130xl version 3.1 DNA sequencer (Applied Biosystems, Foster City, CA, USA). The sequences were analyzed for the presence of mutations using Lasergen program (DNASTAR, Inc., Madison, USA). Multiple sequence alignment was carried out using MegAlign program of DNASTAR and WebLogo was created using online software (http://weblogo.berkeley.edu/WebLogo: A Sequence Logo Generator).
| Results | | |
Genomic DNA was isolated from patients and control blood samples and the integrity was checked by running on 0.8% agarose gel electrophoresis. The purity was checked by A 260 /A 280 and was found to be 1.8, which suggests the good quality of genomic DNA.
All the 20 coding regions including exon-intron boundary of MAP3K1 gene were amplified using gene specific primers and sequenced using automated DNA sequencer. Upon sequence analysis, a total of four different sequence variants including one missense, two silent and one insertion have been identified. The missense mutation (c.2416 G > A p.D806N; rs702689) was identified in exon 14 [Figure 1]a of MAP3K1 gene in four patients (2-4 and 7). The silent mutation (c.3084A > G p.Q1028Q; rs3822625) was also identified in patient 6 present in exon 14 [Figure 1]b; whereas the other silent mutation (c. 1284G > A p.T428T; rs832575) was identified in exon 6 [Figure 1]c in patient 10. Two patients (8 and 10) were found to have insertion mutation (c. 2822_2824insCAA p.T942ins; rs34869245) present in exon 14 [Figure 1]d of MAP3K1 gene.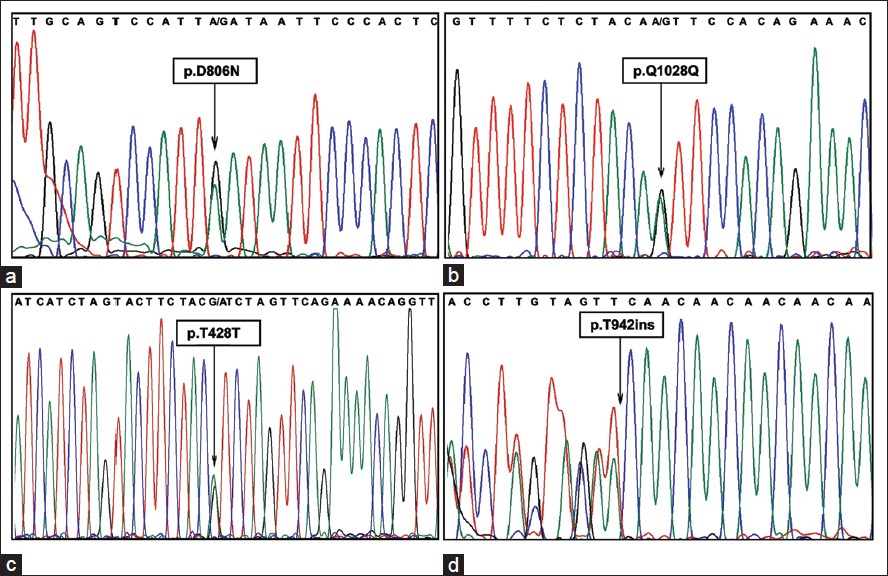 | Figure 1: Deoxyribonucleic acid sequence chromatogram showing the presence of mutations. (a) p.D806N mutation in exon 14 of mitogen activated protein kinase kinase kinase 1 (MAP3K1) gene. This is a heterozygous mutation indicated by an arrow showing presence of two overlapping peaks corresponding to two nucleotides A (wild type) and G (mutant) (b) p.Q1028Q mutation in exon 14 of MAP3K1 (c) p.T428T heterozygous mutation in exon 6 of MAP3K1 (d) p.T942ins mutation in exon 14 of MAP3K1 showing insertion of CAA nucleotide triplet. The chromatogram for this mutation has been reverse complemented for which the frameshift has been depicted toward left
Click here to view |
In-silico analysis of p.D806N missense mutation has been carried out to find out the importance of the residue. Multiple sequence alignment was carried out with its homologs retrieved from NCBI database and those include amino acid sequences of Homo sapiens (Accession No. NP_005912.1), Gorilla gorilla gorilla (XP_004058877.1), Pan troglodytes (XP_003310777.1), Pongo abelii (XP_002815621.1), Ailuropoda melanoleuca (XP_002928301.1), Macaca mulatta (XP_002804414.1), Nomascus leucogenys (XP_003266148.1), Callithrix jacchus (ABY79119.1), Bos taurus (NP_001192835.1), Equus caballus (XP_001916604.1), Canis lupus familiaris (XP_535240.4), Sus scrofa (XP_003134021.1), Felis catus (XP_003981022.1), Gallus gallus (XP_424734.3), Mus musculus (NP_036075.2), Anolis carolinensis (XP_003216255.1), Rattus norvegicus (NP_446339.1), Callicebus moloch (ACA57887.1), Dasypus novemcinctus (ACO15795.1), Monodelphis domestica (XP_001381167.1), Oryctolagus cuniculus (ACJ73998.1), Otolemur garnettii (XP_003782799.1), Taeniopygia guttata (XP_004175671.1) and Rhinolophus ferrumequinum (ACC62498.1). The WebLogo was also created using online software (http://weblogo.berkeley.edu/WebLogo: A Sequence Logo Generator). Both multiple sequence alignment and weblogo revealed that the p.D806N was well conserved across species [Figure 2]a and 2b. The polyphen analysis (http://genetics.bwh.harvard.edu/pph2/) was also carried out and the score for this missense mutation was found to be 0.999. | Figure 2: Multiple sequence alignment with the homologues from different species showing the extent of conservation at the mutation site. (a) A multiple sequence alignment of p.D806N mutation showing the conserved aspartate (D) across species (highlighted by red shaded box) (b) WebLogo showing the conservation of p.D806N mutation showing the conserved aspartate (D) residue indicated by highlighted area
Click here to view |
| Discussion | | |
DSD consist of a wide range of disorders and are more common in those with an XY karyotype. [7] The most widely used method to find out the genetic abnormality in 46,XY DSD cases is by sequencing the exonic region of suspected genes and to analyze those for the presence of mutation/s. In this study, mutation analysis in all exonic regions of MAP3K1 gene was carried out in a cohort of 10 patients including one family with two affected sisters.
Several sex chromosomal and autosomal genes are responsible for sexual development. In our earlier study, two homozygous mutations in the Desert hedgehog (DHH) gene were reported. Two mutations were a homozygous deletion (c. 271_273delGAG) that resulted in a deletion of one amino acid (p.D90del) and a homozygous duplication (c. 57-60dupAGCC) that resulted in premature termination resulting in non-functional DHH protein. Structural studies on the p.D90del mutant revealed that the mutation could seriously perturb the interaction of DHH with its binding partners. [8]
Patients involved in present study were already screened for SRY, SF1 and DHH gene and were found to have no mutations in any of the above genes. MAP3K1 gene is known as a member of the MEK/ERK signaling pathway. MAPK cascades are key signaling pathways involved in the regulation of normal cell proliferation, survival and differentiation. An aberrant regulation of MAPK cascades is known to contribute to cancer and other human diseases. [9] However recently, MAP3K1 was associated with 46,XY DSD in a study where a linkage analysis on long arm of chromosome five was carried out in 46,XY DSD patients from two families and 11 sporadic cases. MAP3K1 expression was also observed throughout the mouse embryonic gonad at 11.5 dpc which is the sex-determining stage of gonad development. Staining was observed within the testis cords at 13.5 dpc in a pattern indicative of Sertoli cell expression. [5] However, despite of observing minor abnormalities in testis development in mice lacking MAP3K1 gene, Warr et al. concluded that MAP3K1 has no significant role for mouse testis determination. [6]
In four of our patients, a missense mutation p.D806N was identified in exon 14 which of caused replacement aspartic acid (D) by aspargine (N) at position number 806 in amino acid sequence. aspartic acid is an acidic amino acid present in the wild MAP3K1 replaced by asparagine, neutral and polar amino acid. Due to change in the polarity, the secondary structure of the protein may be affected. Multiple sequence alignment and Weblogo revealed that the amino acid Aspartate (D) at position 806 is highly conserved across species. This phylogenetically conserved residue indicated the importance of this residue in structure and function of the protein. The pathogenicity of this mutation was tested using polyphen and the score was found to be 0.999, indicating high probability of pathogenic mutation. Genotype-phenotype correlation of this mutation have been carried out and found that all the patients with this mutation had hypospadias with associated gonadal abnormality. However, interestingly this missense mutation was found to be a polymorphism in NCBI SNP database. The significant association of this mutation have been shown to be associated with asthma. [10] However, genital abnormality with this polymorphism has not been reported.
A silent mutation (p.Q1028Q) was identified on exon 14 of MAP3K1 in patient 6. Clinical presentation of this patient had bilateral, well-developed, palpable gonads with ambiguous genitalia. Since, the mutation does not change the amino acid, thus may not affect protein structure.
In another patient, two mutation comprising of one deletion mutation (p.T942ins) and silent mutation (p.T428T) were identified in patient 10. The clinical presentation of this patient was observed like androgen insensitivity syndrome (no mutation in AR gene). In addition, the deletion mutation (p.T942ins) was also present in patients 8.
| Conclusion | | |
We have identified four sequence variants in MAP3K1 gene including one missense mutation, two silent mutations, and a deletion mutation. All the mutations were found to be polymorphism upon comparing to dbSNP. However, in-silico analysis of the missense mutation revealed to be a pathogenic mutation. Further in-vitro analysis would confirm the damaging effect of this mutation. In this report, we reported mutation/polymorphism that may not be directly associated with the sexual development. However, large case-control study is required for establishing the involvement of MAP3K1 in the development of genital ambiguity.
| Acknowledgment | | |
We thank all the patients and their families for their contribution in this study. The authors are also thankful to Director, NIRRH for providing the necessary facilities, the Indian Council of Medical Research (ICMR), Government of India for providing financial grants for the study.
| References | | |
| 1. | Hersmus R, Stoop H, White SJ, Drop SL, Oosterhuis JW, Incrocci L, et al. Delayed Recognition of Disorders of Sex Development (DSD): A missed opportunity for early diagnosis of malignant germ cell tumors. Int J Endocrinol 2012;2012:671209. 
|
| 2. | Ludbrook LM, Bernard P, Bagheri-Fam S, Ryan J, Sekido R, Wilhelm D, et al. Excess DAX1 leads to XY ovotesticular disorder of sex development (DSD) in mice by inhibiting steroidogenic factor-1 (SF1) activation of the testis enhancer of SRY-box-9 (Sox9). Endocrinology 2012;153:1948-58.
|
| 3. | Karandikar M, Xu S, Cobb MH. MEKK1 binds raf-1 and the ERK2 cascade components. J Biol Chem 2000;275:40120-7.
|
| 4. | Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, et al. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr Rev 2001;22:153-83.
|
| 5. | Pearlman A, Loke J, Le Caignec C, White S, Chin L, Friedman A, et al. Mutations in MAP3K1 cause 46, XY disorders of sex development and implicate a common signal transduction pathway in human testis determination. Am J Hum Genet 2010;87:898-904.
|
| 6. | Warr N, Bogani D, Siggers P, Brixey R, Tateossian H, Dopplapudi A, et al. Minor abnormalities of testis development in mice lacking the gene encoding the MAPK signalling component, MAP3K1. PLoS One 2011;6:e19572.
|
| 7. | Ahmed SF, Bashamboo A, Lucas-Herald A, McElreavey K. Understanding the genetic aetiology in patients with XY DSD. Br Med Bull 2013;106:67-89.
|
| 8. | Das DK, Sanghavi D, Gawde H, Idicula-Thomas S, Vasudevan L. Novel homozygous mutations in Desert hedgehog gene in patients with 46, XY complete gonadal dysgenesis and prediction of its structural and functional implications by computational methods. Eur J Med Genet 2011;54:e529-34.
|
| 9. | Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007;26:3291-310.
|
| 10. | Szczepankiewicz A, Sobkowiak P, Rachel M, Brêborowicz A, Schoneich N, Bruce K, et al . Multilocus analysis of candidate genes involved in neurogenic inflammation in pediatric asthma and related phenotypes: A case-control study. J Asthma 2012;49:329-35.
|
[Figure 1], [Figure 2]
[Table 1]
| This article has been cited by | | 1 |
DSDs: genetics, underlying pathologies and psychosexual differentiation |
|
| Valerie A. Arboleda,David E. Sandberg,Eric Vilain | | Nature Reviews Endocrinology. 2014; | | [Pubmed] | [DOI] | |
|
|
|
|










