|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 4 | Page : 483-486 |
| |
An incidental case of dihydropyrimidine dehydrogenase deficiency: One case, multiple challenges
Hamoud H Al Khallaf1, Miao He1, Angela Wittenauer2, Elizabeth E Woolley3, Mariagrazia Cunto2, Muhammad Ali Pervaiz4
1 Department of Human Genetics; Department of Biochemical Genetics; Emory Genetics Laboratory, Emory University, Atlanta, Georgia, USA
2 Department of Human Genetics, Emory University, Atlanta, Georgia, USA
3 Department of Human Genetics; Emory Genetics Laboratory, Emory University, Atlanta, Georgia, USA
4 Department of Hospital Medicine, WellStar Health System, Douglasville, Georgia; Genomic Medicine Consultants, Decatur, GA, USA
| Date of Web Publication | 4-Jan-2014 |
Correspondence Address:
Muhammad Ali Pervaiz
WellStar Health System, 8954 Hospital Drive, Douglasville, Georgia 30134
USA
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.124382

 Abstract Abstract | | |
Dihydropyrimidine dehydrogenase (DPD) deficiency is an autosomal recessive disorder that shows large phenotypical variability, ranging from no symptoms to intellectual disability, motor retardation, and convulsions. In addition, homozygous and heterozygous mutation carriers can develop severe 5-fluorouracil (5-FU) toxicity. The lack of genotype-phenotype correlation and the possibility of other factors playing a role in the manifestation of the neurological abnormalities, make the management and education of asymptomatic DPD individuals more challenging. We describe a 3-month-old baby who was incidentally found by urine organic acid testing (done as part of positive newborn screen) to have very high level of thymine and uracil, consistent with DPD deficiency. Since the prevalence of asymptomatic DPD deficiency in the general population is fairly significant (1 in 10,000), we emphasize in this case study the importance of developing a guideline in genetic counseling and patient education for this condition as well as other incidental laboratory findings.
Keywords: 5-Fluorouracil, dihydropyrimidine dehydrogenase, genetic counseling, newborn screening, public health
How to cite this article:
Al Khallaf HH, He M, Wittenauer A, Woolley EE, Cunto M, Pervaiz MA. An incidental case of dihydropyrimidine dehydrogenase deficiency: One case, multiple challenges. Indian J Hum Genet 2013;19:483-6 |
How to cite this URL:
Al Khallaf HH, He M, Wittenauer A, Woolley EE, Cunto M, Pervaiz MA. An incidental case of dihydropyrimidine dehydrogenase deficiency: One case, multiple challenges. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:483-6. Available from: http://www.ijhg.com/text.asp?2013/19/4/483/124382 |
| Introduction | |  |
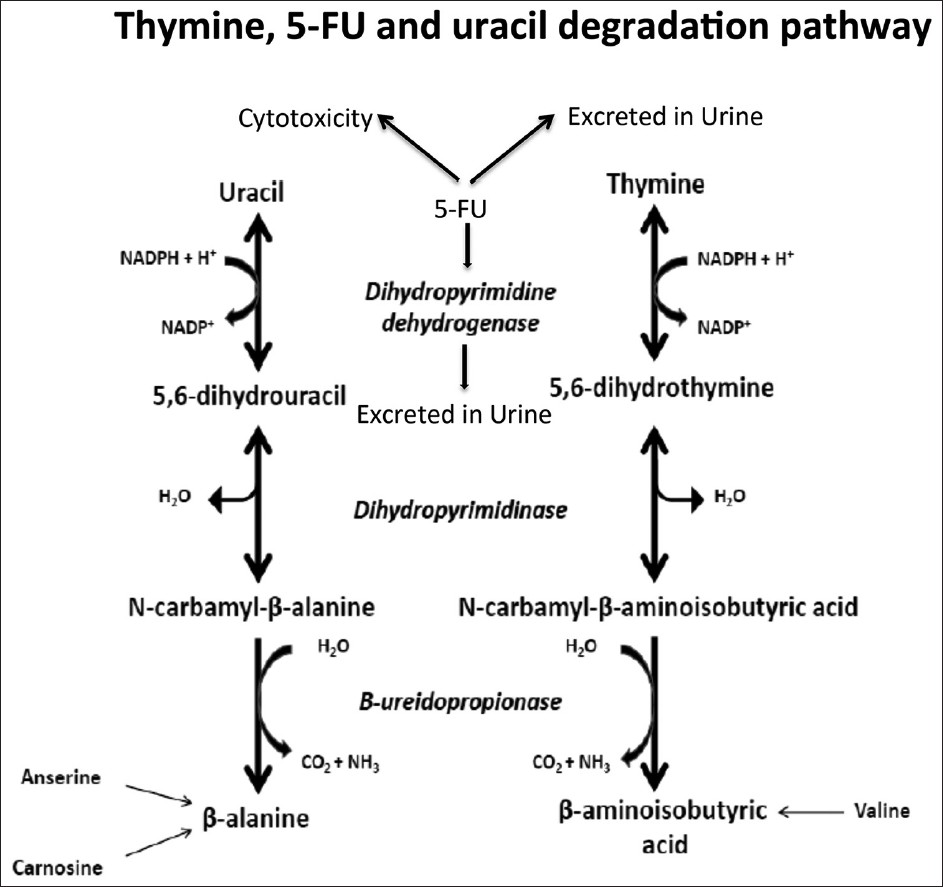
The pathway for the catabolism of uracil and thymine in mammalian livers consists of three consecutive steps [Figure 1]. Dihydropyrimidine dehydrogenase (DPD; EC 1.3.1.2) is the initial and rate-limiting enzyme in that pathway. It catalyzes the reduction of uracil and thymine to 5,6-dihydrouracil and 5,6-dihydrothymine, respectively. In DPD deficiency (OMIM 274270), thymine and uracil accumulate in blood and cerebrospinal fluid resulting in their excess excretion in urine. This infrequently described disease is an autosomal recessive disorder that shows large phenotypical variability, ranging from no symptoms to psychomotor retardation and convulsions. [1]
In addition to its role in thymine and uracil metabolism; DPD plays an important role in the catabolism of >80% of the administered dose of 5-fluorouracil (5-FU) [Figure 1], an antineoplastic uracil analogue. [2] Patients with a partial or complete enzyme deficiency can suffer from severe and potentially lethal toxicity following 5-FU administration. [3] Therefore, reliable identification of DPD deficiency is essential to identify cancer patients at risk.
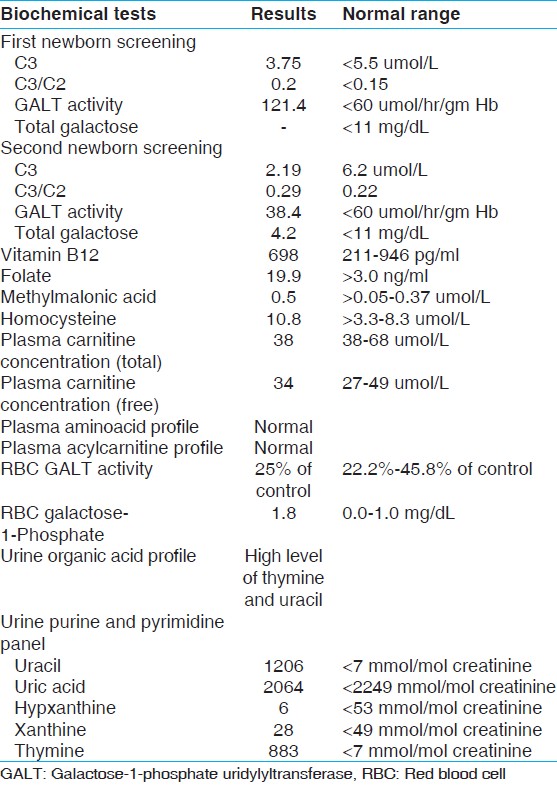
In this case report, we describe an incidental diagnosis of DPD deficiency and the challenges we faced in its management. The proband is a 3-month-old Caucasian boy referred to our metabolic clinic at 1 month and 18 days of age due to a positive newborn screen. The child was a product of full-term pregnancy delivered via spontaneous vaginal delivery. Both parents were Caucasian. There was no consanguinity. This proband had an elder maternal half-brother who was diagnosed with duarte galactosemia with no other medical illness. The first newborn screening of this proband was positive with elevations of C3 (propionylcarnitine) and C3/C2 ratio (propionylcarnitine/acetylcarnitine). However, his second newborn screen was normal for these two parameters, but showed decreased activity of galactose-1-phosphate uridylyltransferase (GALT) enzyme [Table 1]. He also had a history of emesis that improved with Similac Isomil formula. The mother was not vegan or strict vegetarian. She had been taking her prenatal vitamins since the 6 th week of pregnancy. The patient was not microcephalic, had no history of convulsions, motor retardation, or intellectual disability. Other than vomiting, the patient had no other medical issue that could be attributed to galactosemia such as poor weight gain, lethargy, hyptonia, jaundice, or hepatomegaly.
His red blood cell (RBC) galatose-1-phosphate was elevated, while his RBC GALT activity was 25% of the concurrent control [Table 1]. Common mutation analysis of GALT gene detected one copy of the c. 563A > G (p.Q188R) missense mutation. Subsequent sequence analysis confirmed the presence of that mutation. No other mutation; deletion, duplication, or other structural abnormality was found in that gene. His plasma acylcarnitine profile, plasma amino acid profile, plasma carnitine concentration, folate, vitamin B12, and total homocysteine levels were within normal limits. Although his urine organic acid profile did not show increased concentrations of either propionic or methylmalonic acid, the concentration of thymine and uracil were significantly elevated [Figure 2]. Urine purine and pyrimidine analysis confirmed these results [Table 1].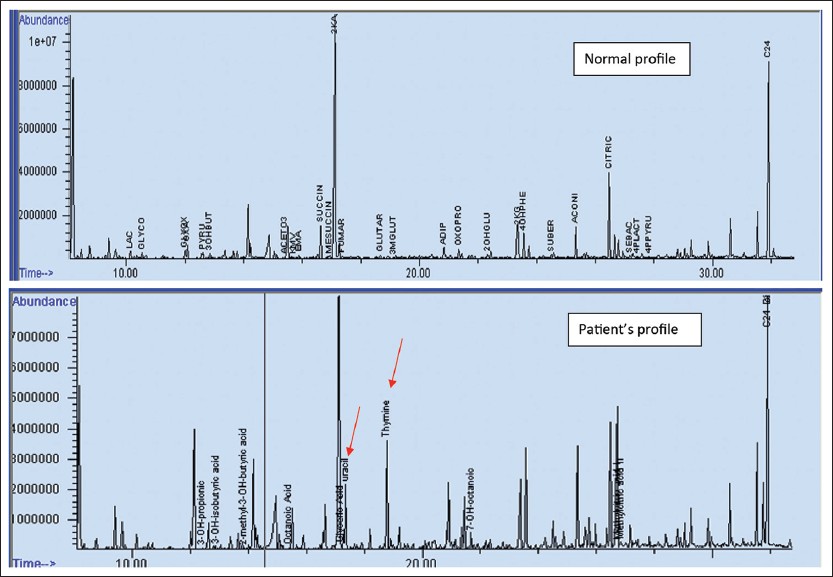 | Figure 2: Normal urine organic acid profile and our patient's profile showing two prominent thymine and uracil peaks (red arrows) in the later
Click here to view |
| Discussion | | |
This case demonstrates the issues, which arise when inadvertent results are seen as part of follow-up of positive newborn screening. [4] While communicating confirmatory test results to the family, the patient's family was also counseled about false-positive screen results arising from newborn screening. The fact that positive newborn screening results always need to be confirmed by diagnostic testing was also explained. These tests provide higher specificity and can help determine a true positive result from a false one. The proband's follow-up confirmatory testing showed increased thymine and uracil in urine which was confirmed quantitatively on the urine purine and pyrmidine panel. These biochemical results were consistent with DPD deficiency, a disease that is not part of newborn screening panel. One of the challenges we faced with this case, was the lack of standardized guidelines on whether such incidental finding should be reported to the family or not. What characterizes this disease is that even those patients with complete DPD enzyme deficiency, have wide spectrum of clinical manifestations, from completely normal to quite severe. [5] This disease variation added more complexity to this case and made the decision to report or not, more challenging. However, the decision was taken to report these results as we wanted to monitor the baby's development as well as wanted the mother to be aware of risk of developing 5-FU toxicity, if a need to use such a drug arises in the future. The fact that approximately 2 million patients receive this drug worldwide each year emphasizes the importance of such a disease. [6] They were also informed that genotype testing is considered one of the confirmatory tests for this disease; however, due to health insurance issues DNA testing was not done. The family was made aware that their child would be regularly followed-up with clinical and biochemical assessment. The importance of educating the child about his disease as he grows up was also emphasized.
As newborn screening is implemented in the developing world, [7] it is pertinent to realize that some of the techniques used to diagnose inborn errors of metabolism may pick up additional disorders. A comprehensive plan needs to be in place with all stakeholders (government, physicians, public health labs, and hospitals) involved in how to manage such incidental findings. Limited healthcare budgets in addition to limited expertise in some areas will complicate these issues.
| Acknowledgement | | |
We thank the patient's family for their patience and cooperation.
| References | | |
| 1. | Van Kuilenburg AB, Haasjes J, Richel DJ, Zoetekouw L, Van Lenthe H, De Abreu RA, et al. Clinical implications of dihydropyrimidine dehydrogenase (DPD) deficiency in patients with severe 5-fluorouracil-associated toxicity: Identification of new mutations in the DPD gene. Clin Cancer Res 2000;6:4705-12. 
|
| 2. | Heggie GD, Sommadossi JP, Cross DS, Huster WJ, Diasio RB. Clinical pharmacokinetics of 5-fluorouracil and its metabolites in plasma, urine, and bile. Cancer Res 1987;47:2203-6.
|
| 3. | Lu Z, Zhang R, Carpenter JT, Diasio RB. Decreased dihydropyrimidine dehydrogenase activity in a population of patients with breast cancer: Implication for 5-fluorouracil-based chemotherapy. Clin Cancer Res 1998;4:325-9.
|
| 4. | Tu WJ, He J, Chen H, Shi XD, Li Y. Psychological effects of false-positive results in expanded newborn screening in China. PLoS One 2012;7:e36235.
|
| 5. | Van Kuilenburg AB, Meinsma R, Beke E, Bobba B, Boffi P, Enns GM, et al. Identification of three novel mutations in the dihydropyrimidine dehydrogenase gene associated with altered pre-mRNA splicing or protein function. Biol Chem 2005;86:319-24.
|
| 6. | Ezzeldin HH, Diasio RB. Predicting fluorouracil toxicity: Can we finally do it? J Clin Oncol 2008;26:2080-2.
|
| 7. | Padilla CD, Therrell BL Jr, Working Group of the Asia Pacific Society for Human Genetics on Consolidating Newborn Screening Efforts in the Asia Pacific Region. Consolidating newborn screening efforts in the Asia Pacific region: Networking and shared education. J Community Genet 2012;3:35-45.
|
[Figure 1], [Figure 2]
[Table 1]
|