|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 4 | Page : 487-490 |
| |
Dyschromatosis universalis hereditaria: Infrequent genodermatoses in India
Hari Kishan Kumar Yadalla, Srivalli Pinninti, Anagha Ramesh Babu
Department of Dermatology, M.V. Jayaraman Medical College and Research Hospital, Hoskote, Bangalore, Karnataka, India
| Date of Web Publication | 4-Jan-2014 |
Correspondence Address:
Hari Kishan Kumar Yadalla
Res: 70, Padma Nivasa, Skin Care Clinic, 3rd Cross MG Extension, HV Halli, Raja Rajeswari Nagar, Bangalore - 560 098, Karnataka
India
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.124383

 Abstract Abstract | | |
Dyschromatosis universalis hereditaria (DUH) is a rare genodermatosis reported initially and mainly in Japan. However, subsequent cases have been reported from other countries. We report a case of DUH in a south Indian woman with a positive family history with cosmetic disfigurement and severe psychological impairment.
Keywords: Dyschromatosis symmetrica hereditaria, dyschromatosis universalis hereditaria, genodermatosis, psychological impairment
How to cite this article:
Yadalla HK, Pinninti S, Babu AR. Dyschromatosis universalis hereditaria: Infrequent genodermatoses in India. Indian J Hum Genet 2013;19:487-90 |
How to cite this URL:
Yadalla HK, Pinninti S, Babu AR. Dyschromatosis universalis hereditaria: Infrequent genodermatoses in India. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:487-90. Available from: http://www.ijhg.com/text.asp?2013/19/4/487/124383 |
| Introduction | |  |
Dyschromatoses are a group of disorders characterized by the presence of small irregular hyper- and hypopigmented macules. It is a spectrum of diseases which includes dyschromatosis universalis hereditaria (DUH), dyschromatosis symmetrica hereditaria (DSH), or acropigmentation of Dohi and a segmental form called unilateral dermatomal pigmentary dermatosis (UDPD). DSH was first reported as a clinical entity by Toyama in 1929. [1] It is characterized by a symmetrical distribution of hyper- and hypopigmented macules on the extremities, especially over dorsa of the hands and feet. In 1933 Ichikawa and Higara described DUH which was essentially the same disorder, but occurring in a generalized as opposed to acral distribution. [2]
| Case Report | | |
A 28-year-old unmarried woman from rural south India presented to us with chief complaints of multiple hyper- and hypopigmented lesions over arms, legs, trunk, and buttocks since she was 3-years-old, she was apparently normal prior to that age. The lesions had started over both the legs and gradually spread upwards towards the thighs. Few lesions had appeared during this time over the hands subsequently spreading upwards to the elbows. Later, lesions developed over the buttocks and trunk also. There was no history of photosensitivity or photophobia. There was no history of handling any chemical directly or any significant history of drug intake. Her mental status was subnormal with depression.
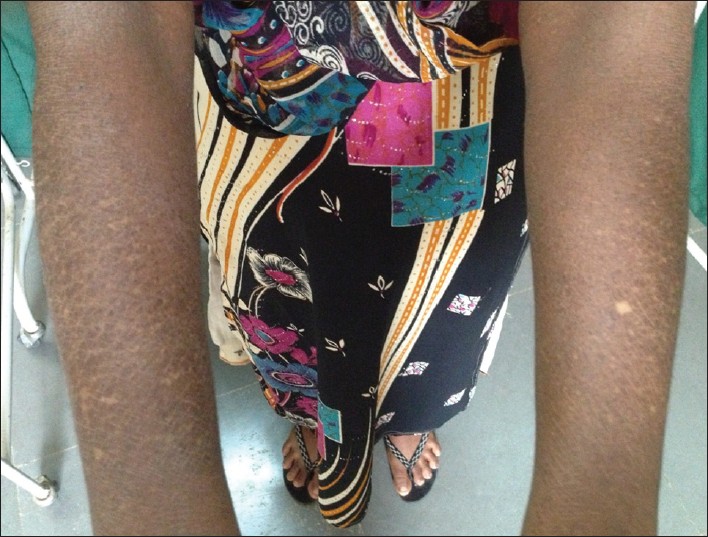
Physical examination revealed numerous asymptomatic, generalized, 0.5-1 cm hyperpigmented macules interspersed with spotty hypopigmented macules. The lesions were dense on the limbs and trunk. Palms and soles were not involved. Her face showed mild involvement. The hair, nails, teeth, and mucosae appeared normal. There was no apparent atrophy, erythema, or telangiectasia [Figure 1] and [Figure 2]. Systemic examination did not reveal any abnormality. Other routine investigations and chest X-ray were within normal limits. Venereal disease research laboratory (VDRL) and human immunodeficiency syndrome test was negative. There was no history of consanguinity among the parents. She reported that her father and sister had similar lesions, but in a milder form. She was depressed since she was not married due to the cosmetic disfigurement and felt worthless, we counseled the patient and also referred her for psychiatric consultation.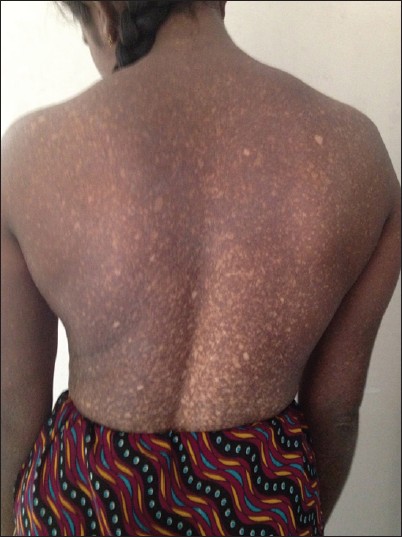 | Figure 1: Generalized, 0.5 - 1 cm hyperpigmented macules interspersed with spotty hypopigmented macules. The lesions were dense on the limbs and trunk with sparing of the palms and soles
Click here to view |
A skin biopsy was taken from both the hyper- and hypopigmented lesions. Section studies shows epidermis in both the biopsies was mildly atrophic with hyperkeratosis. Basal layers showed mild vacuolar change, papillary dermis with melanin incontinence, and dilated dermal vessels. Hyperpigmented lesions showed increase in pigment extending into the stratum spinosum; whereas, in the hypopigmented lesions it showed decrease in pigment. Dermis in both the biopsies showed mild perivascular and periadnexeal lymphomononuclear infitrate [Figure 3] and [Figure 4].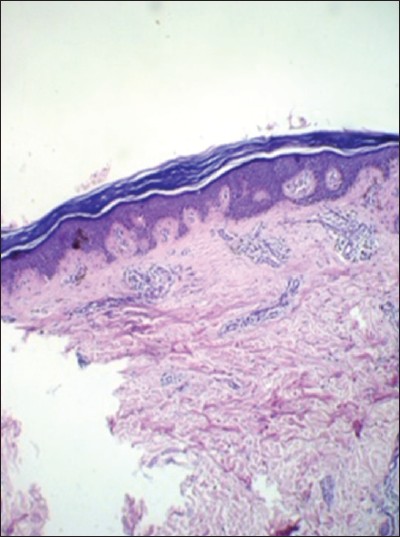 | Figure 3: Histopathology of the skin lesion shows epidermis was mildly atrophic with hyperkeratosis. Basal layers showed mild vacuolar change, papillary dermis with melanin incontinence, and dilated dermal vessels (hematoxylin and eosin (H and E, ×10)
Click here to view |
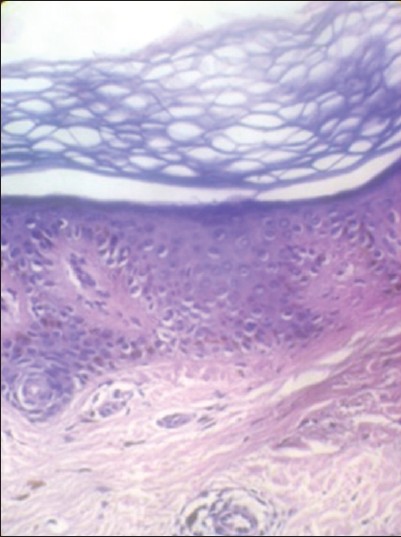 | Figure 4: Histopathology of the skin lesion: In high power, section shows increase in pigment extending into the stratum spinosum. Dermis showed mild perivascular and periadnexeal lymphomononuclear infitrate (H and E, × 10)
Click here to view |
The patient is currently being treated with narrowband ultraviolet B (NBUVB) therapy, has completed 20 sessions and feels her lesions have improved. Also, was put on escitalopram 10 mg in the morning and clonazepam 0.5 mg at night for depression. Patient feels after the counseling and with treatment, is happier with the cosmetic appearance of the lesions and has started working too.
| Discussion | | |
DUH is a rare genodermatosis which has been reported most often from Japan. Although majority of cases show autosomal dominant pattern of inheritance, a few have inherited it in an autosomal recessive fashion. The etiology of this disorder is not yet known. A variable autosomal mode of inheritance of DUH has also been described. In our case, patient's father and her sister had similar complaints in a milder form.
Two major types have been described, based on distribution of lesions as follows: DUH, first reported in 1933 by Ichikawa and Higara, [1] in which mixtures of hyper- and hypopigmented macules occur all over the body; and DSH, also known as acropigmentation of Dohi, described in 1929 by Toyama, [2] which is a localized form involving only the acral areas. Both conditions are seen most commonly in Japan. However, a few cases of DUH were described among Europeans, South Americans, Indians, and Saudi Arabians and in Tunisia. [3],[4],[5],[6],[7],[8],[9]
In DUH, skin lesions are usually present in the first years of life. The trunk and extremities are the dominant sites. Facial lesions were seen in almost 50% of affected individuals, but involvement of palms and soles is unusual. [6] Abnormalities of hair and nails have also been reported. [7] The histopathology typically shows a focal increase or decrease in melanin content of the basal layer (depending on the type of the lesion biopsied) and occasionally pigmentary incontinence. In a recent ultrastructural skin investigation, Nuber et al. indicated that DUH is a disorder of melanosome synthesis rate or in melanocyte activity and not a disorder of melanocyte number. [10]
Of the 37 previously reported cases from Japan, 15 were men and 22 were women. Eighty-two percent of patients had clinical symptoms before the age of 6 years. Our patient gave a history that her lesions presented at the age of 3 years. DUH may be associated with abnormalities of dermal connective tissue, nerve tissue, or be associated with other systemic complications. [11],[12] No such features characterized our patient.
Lesions of DSH have to be differentiated from xeroderma pigmentosum, since in both the disorders patients clinically show lesions in the photoexposed areas. However, in DUH lesions occur in the unexposed sites as well. Moreover, the lesions show no atrophy or telangiectasia. The lesions also run a benign course. Generally, DUH does not progress or worsen with age. [5] In all reports there was no spontaneous regression. Other differential diagnosis of DUH to be considered are DSH, dyskeratosis congenital, generalized Dowlinge-Degos disease, incontinentia pigmenti (Bloch-Sulzberger), Naegelie-Franceschetti-Jadassohn syndrome, chronic arsenic toxicity, and chronic radiodermatitis. [10]
Once thought to be a disease confined to Japanese, DUH is being reported with increasing frequency in other races as well. From India there have been very few case reports in the past. [5],[6],[7],[8] Despite its rarity it assumes significance as it forms an important differential diagnosis of xeroderma pigmentosum.
These patients often suffer from depression because of cosmetic disfigurement so simultaneous counseling and psychiatric consultation can help them lead more fulfilling and productive lives. Role of NBUVB as a treatment modality should be evaluated and tried for better cosmetic appearance.
| References | | |
| 1. | Ichikawa T, Higara Y. About a pigmentary anomaly unprecedented. Jpn J Dermatol 1933;34:360-4. 
|
| 2. | Toyama I. Dyschromatosis symmetrica herediteria. Jpn J Dermatol 1929;29:95-6.
|
| 3. | Al Hawsawi K, Al Aboud K, Ramesh V, Al Aboud D. Dyschromatosis universalis hereditaria: Report of a case and review of the literature. Pediatr Dermatol 2002;19:523-6.
|
| 4. | Bukhari IA, El-Harith EA, Stuhrmann M. Dyschromatosis universalis hereditaria as an autosomal recessive disease in five members of one family. J Eur Acad Dermatol Venereol 2006;20:628-9.
|
| 5. | Merino de Paz N, Rodríguez-Martin M, Contreras Ferrer P, Pestana-Eliche M, Martin-Herrera A, Noda-Cabrera A. Photoletter to the editor: Dyschromatosis universalis hereditaria: An infrequently occurring entity in Europe. J Dermatol Case Rep 2012;3:96-7.
|
| 6. | Rai R, Kaur I, Handa S, Kumar B. Dyschromatosis universalis hereditaria. Indian J Dermatol Venereol Leprol 2000;66:158-9.
[PUBMED]  |
| 7. | Naik CL, Singh G, Rajashekar TS, Okade R. Dyschromatosis universalis hereditaria. Indian J Dermatol 2009;54:74-5.
|
| 8. | Kenani N, Ghariani N, Denguezli M, Sriha B, Belajouza C, Nouira R. Dyschromatosis universalis hereditaria: Two cases. Dermatol Online J 2008;14:16.
|
| 9. | Sethuraman G, Srinivas CR, D'Souza M, Thappa DM, Smiles L. Dyschromatosis universalis hereditaria. Clin Exp Dermatol 2002;27:477-9.
|
| 10. | Nuber UA, Tinschert S, Mundlos S, Hauber I. Dyschromatosis universalis hereditaria: Familial case and ultrastructural skin investigation. Am J Med Gen A 2004;125A: 261-6.
|
| 11. | Miyamura Y, Suzuki T, Kono M, Inagaki K, Ito S, Suzuki N, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet 2003;73:693-9.
|
| 12. | Suzuki N, Suzuki T, Inagaki K, Ito S, Kono M, Fukai K, et al. Mutation analysis of the ADAR1 Gene in Dyschromatosis Symmetrica Hereditaria and genetic differentiation from both Dyschromatosis Universalis Hereditaria and Acropigmentatio Reticularis. J Invest Dermatol 2005;124:1186-92.
|
[Figure 1], [Figure 2], [Figure 3], [Figure 4]
|