Stress Responsivity, Addiction, and a Functional Variant of the Human Mu-Opioid Receptor Gene
Addictions may be defined as a state of drug hunger or craving leading to compulsive drug-seeking and drug-taking behaviors, without regard to potential or actual negative consequences. There are at least three different categories of factors that contribute to the vulnerability of developing a specific addiction, once self-exposed (1). The first category includes a myriad of environmental factors, including prenatal and perinatal events; events occurring in early childhood; and later events, such as peer pressure, cues, conditioning, setting for drug self-exposure, and concomitant ongoing psychiatric diagnosis, such as depression or anxiety. External stressors (and the stress they cause) are another important environmental factor contributing to both the development of, and relapse to, addictions. The second category of factors that contribute to the development of addiction includes drug-induced factors. Each of the short-acting drugs of abuse (e.g., cocaine, morphine, etc.), when administered to or self-administered in animals in a mode mimicking the human pattern of abuse, leads to a variety of molecular neurobiological changes, including changes in gene expression, protein concentrations, and synaptogenesis and, in turn, to altered behaviors. The third category of factors is genetic factors. A heritable basis for addictions has been firmly established by many human genetic family studies, in which the influence of family environmental, non-family environmental, and genetic factors can be differentiated. In one important study of more than 3,000 twin pairs from the Vietnam Era Twin Registry, Tsuang and colleagues reported that both environmental and genetic factors influenced abuse and dependence for several types of drugs, with genetic factors accounting for over 50% of the variance for opiate abuse or dependence (2). In addition to a common or shared genetic vulnerability that influenced all drugs studied, they also identified genetic factors that were specific to each drug category. The greatest degree of drug-specific genetic influence was found for abuse or dependence on opiates (1, 2). Each category of factors influences both the initial and early perception of a self-administered drug and contributes also to the progression from initial and occasional use, to intermittent use, and on to regular use and addiction, or, alternatively, to early cessation of any drug use. A variety of personality factors and traits may also contribute to initiation of drug abuse, including impulsivity and risk-taking, as well as intrinsic atypical stress responsivity (1).
Opiate and opioid drugs mediate their analgesic and rewarding properties through the mu (μ)-opioid receptor. In 1994, in collaboration with Yu and colleagues, we strategized to look for sequence variants within the coding region of the μ-opioid receptor gene and studied both healthy volunteer subjects and former severe heroin addicts undergoing methadone maintenance treatment. These studies were conducted primarily in three ethnic groups: Caucasians, African Americans, and Hispanics. We identified several variants; strikingly, two were of high overall allelic frequency in the mixed populations studied. The C17T variant results in an amino acid change of alanine to valine at position 6 (Ala6Val or A6V) (overall allelic frequency 6.6%), and the A118G variant, which results in an amino-acid change from asparagine to aspartic acid at position 40 (Asn40Asp or N40D) (overall allelic frequency 10.5%). Both these substitutions are in the N-terminal extracellular domain of the receptor (Figure 1⇓). Each variant was present in significantly different allelic frequencies among the three ethnic groups. While we were proceeding to conduct molecular and cellular studies to assess functional consequences of the A118G variation, two groups reported genotyping of the μ-opioid receptor gene in studies of addictive disorders. One identified only the C17T variant; the second, the A118G variant only (3). In the study that identified the A118G variant, no association of the μ-opioid receptor variant was linked to alcoholism. Subsequently, we identified these two (and a few less common variants) and characterized in vitro the functional differences between the receptors encoded by the two different alleles (4). Radioligand binding assays of cell lines stably transfected with the 118G variant or the more common prototypic 118A receptor revealed that the two different receptors bound most ligands (including a wide variety of exogenous opiates and opioids) with similar affinity. Similarly, there was no difference in binding affinity of the synthetic peptide Tyr-d-Ala-Gly-MePhe-Gly-ol (DAMGO), endogenous met-enkephalin, or endomorphins 1 and 2 to the two receptors. However, when the longest of the endogenous opioids, β-endorphin, derived from the single gene product proopiomelanocortin was tested, we found striking differences. β-Endorphin bound with a three-fold higher binding affinity (Kι) to the 118G variant receptor as compared to binding to the prototype receptor. Also, in Xenopus oocytes expressing either the variant or prototype receptor coexpressed with G protein–activated inwardly-rectifying potassium channels (GIRKs), a three-fold greater channel activation was found when β-endorphin activated the 118G allele receptor as compared to activation of the prototype receptor (4) (Figure 2⇓). Significantly, the 118G variant abolishes a putative site of N-glycosylation in the extracellular domain of the receptor. During the production of the stable cell lines for the ligand binding assays, the receptor resulting from the A118G variant was usually expressed in much lower amounts than was the prototype receptor, adding to the complexity of determining binding affinities. Based on the above findings, the 118G variant receptor was thought functionally different than the prototype receptor in vivo. Endogenous opioids, and β-endorphin in particular, are widely distributed in the central nervous system and in the periphery. Because β-endorphin participates in many physiological systems and is produced in the pituitary, immune cells, gastrointestinal tract, and in several regions within the brain, we hypothesized that the variant receptors resulting from the A118G polymorphism would lead to alterations in normal physiology, as well as a strong likelihood of association with specific addictive diseases (4, 5). Thus, research was undertaken to discern a possible role for the μ-opioid receptor 118G variant in the normal variations of stress response and other aspects of human physiology, including the possible association of this variant with opiate addiction and alcoholism.
Wand, Kranzler, and their colleagues (6–8) observed the effects of administering the μ-opioid receptor–preferring antagonist nalox-one to healthy volunteers with no history of any addictive disease of any type. Subjects were genotyped for the A118G polymorphism. Each of these studies had quite a small number of subjects, but each found a significantly greater serum cortisol response, indicating greater activation of the hypothalamic-pituitary-adrenal (HPA) axis following intravenous naloxone, and thus a greater response to this chemically-induced stressor in subjects carrying the 118G allele. Additionally, in a stress-minimized setting, basal amounts of cortisol in subjects with the 118G allele were significantly higher than in volunteers with the prototype 118A allele, suggesting there is modest dysregulation of the HPA axis, although the actual levels of the hormone cortisol did not exceed the upper limit of normal values, as defined in clinical medicine (9).
In another study, the effect of naltrexone vs placebo on craving, alcohol self-administration, and activation of the stress-responsive HPA axis was conducted in a group of alcoholics (10). Findings from that study provide direct support for the hypothesis that alcoholics are seeking, in part, a modest activation of the HPA axis, and when that is achieved, they will stop drinking. Because informed consent providing specific consent for genetics studies was not obtained, genotyping could not be performed. In a second study from our laboratory, also without allelic profiling of the μ-opioid receptor, the effects of a single oral dose of naltrexone on HPA axis activation was studied in healthy young adults, half of whom (eight) were family history–positive for alcoholism, whereas the other half (seven) were family history–negative. Although the overall group showed an activation of the HPA axis following a single oral dose of naltrexone, subsequent analysis showed that activation of the HPA axis in those with a positive family history of alcoholism was greater than in those with the negative family history (11). A third study, involving male heavy drinkers, observed the impact of neutral cue vs alcohol cue reactivity in the sequence of three-minute trials (12). After the three-minute cue of alcohol (beer) versus water, those with the 118G variant were significantly more responsive to cue-induced craving following the alcohol (beer) cue.
In an exciting study that provided the first proof-of-principle for pharmacogenetic effects of the A118G variant, two research groups that had conducted several trials on the use of naltrexone for management of alcoholism, but had not consented subjects for genetic profiling, invited all subjects back for genetic restudy (13). Approximately one in six consented (total of 82 subjects), and this data was analyzed according to the original criteria for a positive response to naltrexone. A significant correlation was identified for the presence of one or two copies of the 118G allele with a positive response to naltrexone treatment for alcoholism.
Several studies in populations with relatively low admixture, that is, relatively homogeneous, with limited intermarriage between or among different ethnic, cultural or national groups, point to the 118G allele of the μ-opioid receptor as a risk factor for both alcohol and opiate addictions. In a sample from central Sweden, we identified a positive association of the 118G allele with alcoholism (14). Nichizawa and colleagues also reported association of the 118G allele with alcohol dependence in a Japanese population (15). In another study based on subjects from central Sweden, a highly significant association of the 118G allele with heroin addiction was identified, with an attributable risk of 21% for Swedish subjects whose parents were both Swedish (16). Similarly, Stadlin and colleagues reported a higher prevalence of the 118G allele in heroin addicts than in control subjects in Han Chinese with both parents also of Han descent (17). Although functional variants of many genes may contribute to addiction risk, the μ-opioid receptor A118G polymorphism is clearly emerging as a prime candidate, although not all studies have identified associations (1, 5, 18). Studies of pain and responses to opioid analgesics also point to this gene––and the A118G variant specifically––as a significant determinant of endogenous opioid system functioning and of analgesic and other physiological responses to opioid drugs (18–20).
The μ-opioid receptor belongs to the superfamily of G protein–coupled receptors, is linked to inhibitory Gi/Go proteins, and its actions are mediated through several intracellular signaling pathways (Figure 2⇓). Activation of the μ-opioid receptor results in the inhibition of neuronal firing, but μ-opioid agonists also activate mesocortolimbic dopaminergic signaling through disinhibition of GABAergic interneurons in the ventral tegmental area (VTA). Increased dopamine concentrations, as well as direct opioid effects on the nucleus accumbens and other reward regions of the brain, are known to play a key role in addictions. Our early functional studies led us to the hypothesis that alteration of receptor glycosylation and presentation in the cell membrane would underlie the differences in observed function identified later in clinical studies (1, 4, 5). Recently reported in vitro experiments and an analysis of post-mortem human brains indicate that the 118G-encoded receptors are expressed in lower amounts than those encoded by the prototypic gene (21–23). Whether this difference in expression arises from allelic imbalance––such as differential transcription from one allele compared to another (22), or because of differences in receptor glycosylation (23)––has yet to be fully elucidated. However, in vitro studies using transfected variant receptors encoded by the 118G polymorphism indicate that receptor expression, agonist-induced responses to adenylyl cyclase activity, GIRK channel activation, and also activation of N-type Ca2+ channels are likely to be altered by the polymorphism (2, 22–24).
There is clear evidence now that the functional 118G variant of the μ-opioid receptor gene, which changes an amino acid in the N terminus and leads to greater binding of β-endorphin and enhanced signal transduction, alters at least one specific component of normal physiology (i.e., stress responsivity) and is associated with two addictions, when studied in populations that are not highly admixed. It should be emphasized, however, that no single gene variant will be identified as the sole determinant of addictions––opiate, alcohol, or otherwise. Multiple variants of multiple genes acting in combination contribute to the vulnerability of, or protection from, developing an addiction. Nonetheless, because this variant also causes “physiogenetic changes” (Box 1), and alters responses to μ-opioid antagonists, it may soon be clinically relevant to assess the genotype of the A118G polymorphism in individuals receiving an opioid antagonist for any indication. For example, patients requiring treatment of gastrointestinal hypomotility on an opiate-induced or idiopathic basis may be prescribed an orally administered opioid antagonist with no systemic (or CNS) bioavailability, as first defined by our laboratory (25). We predict that those with the 118G variant will have better response to such treatment. Further, there is already preliminary evidence that alcoholics with one or two copies of the 118G variant are more likely to respond to opioid antagonist treatment. We know that many former heroin addicts with the 118G variant are responding satisfactorily to long-term methadone maintenance treatment, with normalization of their stress-responsive HPA axis, which becomes profoundly disrupted during cycles of addiction. However, we do not know yet whether the length of time or dose in pharmacological treatment to achieve normalization differs based on genotype at the μ-opioid receptor. In the future, primary prevention may include genotyping for variants known to cause functional differences that contribute to addictive diseases. Molecules designed to increase or modestly decrease activity at the μ-opioid receptor when the 118G variant is present, but which have no differential effects for the prototype receptor, could be of importance.
Physiogenetics
In 2000, we coined the term “physiogenetics,” mimicking the long-standing term of “pharmacogenetics.” The term pharmacogenetics had been used for many years, and was originally based on careful clinical observations of significantly different responses to specific pharmacological agents in individuals and in some family members, long before the cloning and sequencing of the human genome. More recently, the specific gene variants involved in pharmacogenetic differences, generally based on drug metabolism or disposition, have been identified. We define the term “physiogenetics” as a difference in response to an individual’s own hormones, neurotransmitters, or other biologically active components owing to a gene variant yielding a different receptor, peptide, or enzyme, or by altering some neurobiological or metabolic pathway.
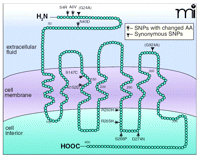
The seven-transmembrane domain G protein–coupled human μ-opioid receptor. Circles represent amino acids. The positions of naturally occurring genetic variants in the coding region, all of which are single nucleotide polymorphisms (SNPs) are indicated, including those that result in an amino acid change and those that do not (synonymous SNPs). The most common coding region variant is the A118G polymorphism, which encodes the substitution of asparagine to aspartic acid (Asn40Asp or N40D) in the extracellular N-terminal domain. This change abolishes one of five putative N-linked glycosylation sites, results in physiogenetic and pharmacogenetic alterations in physiological systems in which the μ-opioid receptor plays a role, and is associated with specific addictions, including alcohol and opioid. In vitro studies have also found alterations in receptor function for several other SNPs, particularly those in the third intracellular loop; however, these variants are rare, and therefore will not figure significantly in common disorders such as addictions. Figure reproduced from (5). ©2000, used with permission from Elsevier.
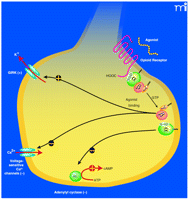
Intracellular signaling pathways of the μ-opioid receptor. Opioid receptors are coupled to inhibitory Gi/Go heterotrimeric G proteins. Agonist binding to the receptor induces an exchange of GDP to GTP on the G protein which dissociates from the receptor; the alpha subunit also dissociates from the β/γ subunits. The GTP-bound α subunit inhibits the enzyme adenylyl cyclase (AC) leading to a decrease in intracellular cyclic AMP (cAMP) concentrations. The β/γ subunits activate G protein–activated inwardly rectifying potassium (GIRK) channels, and inhibit voltage-sensitive calcium channels. Each of these effector systems act to reduce neuronal excitability following agonist activation of the μ-opioid receptor. In vitro studies suggest that signal transduction through each of these pathways is altered by the A118G polymorphism of the μ-opioid receptor gene. Figure modified from (26). ©2001, used with permission from The American Physiological Association.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

K. Steven LaForge, PhD, is a Finnish Academy Research Fellow and Senior Scientist at the Finnish Genome Center of the University of Helsinki in Helsinki, Finland and Visiting Scientist in the Laboratory of the Biology of Addictive Diseases at The Rockefeller University in New York City. He received his BA from the College of Arts and Sciences of Cornell University in Ithaca, New York, and his PhD in pharmacology from Uppsala University in Uppsala, Sweden. His current research program is focused on human studies of genetic influences on pain perception, opioid analgesia, and specific addictions. E-mail steven.laforge{at}helsinki.fi

Mary Jeanne Kreek, MD, is a graduate of Wellesley College and also of the Columbia University College of Physicians & Surgeons, where she received the MD degree. Dr. Kreek joined The Rockefeller Institute in 1964, and with the late Drs. Vincent P. Dole and the late Dr. Marie Nyswander, who also joined the team at that time, performed the initial studies of the use of a long-acting opioid agonist, methadone, in chronic management of heroin addiction. Dr. Kreek is Patrick E. and Beatrice M. Haggerty Professor and Head of the Laboratory of the Biology of Addictive Diseases, at The Rockefeller University, and Senior Physician of The Rockefeller University Hospital. She has received several awards for her scientific research, including the R. Brinkley Smithers Distinguished Scientist Award and Lecture of ASAM, the Betty Ford Award from AMERSA, and the Marian Fischman Award and the Nathan B. Eddy Memorial Award for Lifetime Excellence in Drug Abuse Research, both presented by the College on Problems of Drug Dependence. In 2000, she was conferred with the Doctor Honoris Causa by the University of Uppsala, Sweden and was made a Fellow of the New York Academy of Sciences. In 2004, Dr. Kreek was awarded the Columbia University College of Physicians & Surgeons Alumni Association’s Gold Medal for Lifetime Distinguished Achievements in Academic Medicine. E-mail kreek{at}rockefeller.edu; fax 212 327-8574.

