Transactivation of the EGF Receptor: Is the PDGF Receptor an Unexpected Accomplice?
The transactivation of the cytoplasmic tyrosine kinase domain of the epidermal growth factor receptor (EGFR) by heterologous signals has become an emerging theme in the complex field of receptor-mediated signal transduction (1). Evidence for both direct and indirect EGFR transactivation mechanisms has been proposed, thereby expanding the traditional view of highly specific receptor–ligand interactions. In fact, current data now suggest a wealth of signals impinging on the EGFR, making this receptor a central player in cellular responses.
Initially, the EGFR was found, using gradient centrifugation and chemical cross-linking techniques, to form specific, ligand-dependent, homodimers (2, 3). Several years later the concept of heterodimers between EGFR family members was promulgated and proven (4). This mechanism adds complexity to the family’s signaling output. EGFR and HER2–4 can be detected in heterodimer complexes, each pair activated by one of multiple ligands in the EGF and neuregulin families. HER2, the one family member without a true direct ligand is the most promiscuous and the preferred heterodimerization partner of every other family member. By heterodimerizing with EGFR, HER3, or HER 4, HER2 can alter their signaling output and duration (5, 6). Biological proof of the concept of heterodimerization was gained from mice deficient in HER2, HER4, or their ligand heregulin, which produced the same embryonic lethal phenotype: a failure in a specific step of cardiac development (7, 8).
However, recent data have produced another surprise. The EGFR itself responds to a host of signals from outside the receptor-ligand family (1). EGFR transactivation has been detected using a wide variety of G protein–coupled receptor agonists, phorbol esters, cytokines, chemokines, estrogen, and cell stress signals (1, 9). When tyrosine becomes phosphorylated by these alternative pathways, EGFR behaves in a manner indistinguishable from that of activation by exogenous addition of EGF family ligands (Figure 1). EGFR transactivation has also been demonstrated through other receptor tyrosine kinases, including the insulin-like growth factor-1 receptor (IGF1R) and the platelet-derived growth factor receptor beta (PDGFβR) (10, 11). Thus, crosstalk between receptor tyrosine kinase families provides yet another mechanism for diverse stimuli to tap into unique EGFR signaling and associated mitogenic pathways.
Saito et al. highlight the potential role the PDGFβR tyrosine kinase (11) plays in EGFR transactivation, and suggest a novel mechanism for these effects—there is historical precedence for these findings. Over a decade ago, PDGF was shown to stimulate the phosphorylation of serine residues on the EGFR. This effect, termed “transmodulation,” serves to decrease the binding affinity of EGF to EGFR (3, 12, 13). More recently, PDGFβR and EGFR were isolated from caveolae-containing membrane fractions, providing more evidence of “guilt by association” (14, 15). Also, Habib et al. demonstrated a ligand-independent association of EGFR and PDGFβR when both were transfected into Cos-7 cells (16). Interestingly, these researchers reported EGF-stimulated increases in PDGFβR tyrosine phosphorylation, in addition to the PDGFβR-dependent EGFR phosphorylation shown by Saito et al. and others (11).
However, the first demonstration of functional EGFR-PDGFβR receptor cooperation was observed using murine B82L fibroblasts (17). The effects of PDGF on fibronectin-induced migration were enhanced by coexpression of a kinase-competent EGFR. Enhanced motility correlated with PDGF-stimulated EGFR tyrosine phosphorylation and was prevented by expression of a catalytically inactive or truncated EGFR. This PDGF-dependent EGFR activation was necessary to increase cellular motility. Similarly, He et al. showed that the PDGF-dependent stimulation of the p21-activated kinase (PAK) also required a functional EGFR (18). Selective inhibition of the EGFR (with the chemical inhibitor AG1478) or the use of EGFR-deficient cells abolished the activation of PAK, indicating that PAK may be responsible for PDGF-dependent, EGFR-induced cell motility.
The current study of Saito et al. extends those observations showing parallel, PDGF-stimulated phosphorylation of both PDGFβR and EGFR in vascular smooth muscle cells (11). The investigators used crosslinking reagents to stabilize and detect PDGFβR–EGFR “heterodimers;” this heterologous receptor interaction was ligand- and phosphorylation-independent. Surprisingly, whereas PDGF triggered EGFR tyrosine phosphorylation, the kinase activity of PDGFβR was dispensable for this effect. Incubation with antioxidants [e.g., N-acetylcysteine or Tiron (4,5-dihydroxy-1,3-benzenedisulfonic acid)] or a Src inhibitor (e.g., PP2) disrupted the constitutive PDGFβR–EGFR association, suggesting there may be unidentified factors involved in mediating heterologous receptor association. Lastly, inhibition of PDGF-dependent EGFR transactivation resulted in a partial diminution of extracellular signal-regulated kinase (ERK) activation, strengthening the biological relevance of EGFR transactivation.
The process by which EGFR is transactivated is potentially very important with regard to normal physiology and carcinogenesis; however, the mechanisms of transactivation are numerous, and there are many unresolved issues regarding hormone, cytokine, and PDGF-dependent EGFR activation. The model proposed by Saito and coworkers suggest that a Src- or reactive oxygen species-(ROS)-dependent constitutive heterodimerization of the PDGFβR–EGFR exists and facilitates PDGF-stimulated increases in EGFR tyrosine phosphorylation (Figure 2A⇓). However, although the data on PDGF, Src, or ROS-dependent transactivation of the EGFR and the biological consequences appear firm (11), formal acceptance of constitutive, specific EGFR–PDGFβR heterodimers requires additional experimental proof. The coexistence of PDGFβR and EGFR in caveolae-controlling microdomains, their association with cytoskeletal elements, and the chemical nature of the cross-linking experiments allow for the possibility that the complex is large, indirect, and may depend upon other non-receptor proteins (Figure 2B⇓). These caveats leave open the specificity of PDGFβR–EGFR association but not the transactivation consequences.
The fact that transactivation required PDGF but not the kinase activity of PDGFβR is fascinating. How does the PDGFβR affect EGFR in the absence of its kinase activity? The authors suggest that pre-existing PDGFβR–EGFR heterodimers are present. Does PDGF simply facilitate EGFR homodimerization and autophosphorylation in large receptor oligomers? The involvement of Src or the antioxidant-sensitive step in this process may provide another explanation. The authors suggest that active Src is required for maintenance of the PDGFβR–EGFR heterodimer, possibly through site-specific EGFR phosphorylation. However, this would require a constant level of receptor phosphorylation in the unliganded state. Others have identified EGFR Tyr845 as the site of Src-activating stimuli in B82L fibroblasts (19). Therefore, instead of Src maintaining heterodimers between two different receptor tyrosine kinase families, perhaps PDGFβR, with its known interaction of Src, can be induced to activate Src-mediated EGFR tyrosine phosphorylation without the requirement for PDGFβR tyrosine kinase activity. In other words, PDGFβR homodimers, even without tyrosine kinase activity, might direct Src to EGFR. Antioxidants may prevent ROS-dependent Src activation. Ultimately, experiments involving kinase-inactive versions of the PDGFβR, Src−/− cells, or other chemical techniques, will provide additional means by which to examine putative PDGFβR–EGFR heterodimers and insight into the mechanism of this complex and important regulatory schema. In addition, other kinases must still be considered as indirect participants in EGFR transactivation by PDGF.
Other indirect routes of EGFR activation have been identified. One prevailing model for EGFR transactivation by G protein–coupled receptors or other signals involves a phosphatidylinositol-3' kinase (PI3K), Src, or calcium-dependent tyrosine kinase- (CADTK, or Pyk2)-dependent activation of an ADAM family metalloproteinase (20, 21). These membrane-based metalloproteinases can cleave membrane-anchored growth functions including the EGFR ligands heparin bound-(HB)-EGF and tumor growth factor alpha (TGF-α). The ligand is released from its membrane anchor and thereby provides autocrine or paracrine activation of the EGFR (22–24) (Figure 1⇓).
So what is the motive for this seemingly indiscreet collaboration—EGFR transactivation by multiple signals? As pointed out by Li et al., the combination of these signals may facilitate unique aspects of mitogenesis and migration. Because the EGFR contains a specific actin-binding motif not found in the PDGFβR (25, 26), coactivation of these EGFR-containing heterodimeric receptors may enhance the ability of PDGF to mediate cell motility. In addition, the cooperative PDGFβR–EGFR–dependent activation of PAK may further modulate actin dynamics that are necessary for cell migration (18). Lastly, many experiments in the “early days” of growth factor research indicated that cells need twelve to twenty-four hours of serum stimulation in order to traverse the cell cycle (27–29). In defined cell culture media that contain more physiologically relevant doses of growth factors, a requirement for distinct signals from each of the PDGFβR, EGFR, and IGFR was demonstrated for cell cycle traversal. However, high doses of PDGF can overcome the need for EGF. Perhaps EGFR transactivation by one of the multiple mechanisms noted above short-circuits the need for exogenous EGF ligands (see Figures 1 and 2⇓⇓). Whatever the mechanism, the phenomenon of EGFR transactivation has been reported in some cell systems and is likely to have a basis in the physiology of growth control, tissue formation, or stromal epithelial interaction. Whatever the purpose (and mechanism), further investigations will no doubt result in other intriguing relations.
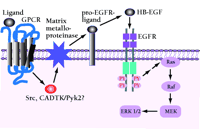
Signaling model of EGFR transactivation by crosstalk with a G protein-coupled receptor (GPCR). A mechanistic signaling model is described for heterologous GPCR coupling to an EGFR-dependent activation of the Ras/mitogen-activated protein kinase (MAPK) pathway (1). GPCR ligand-specific binding activates GPCR-Src- or calciumdependent tyrosine kinase- (CADTK, or Pyk-2) -dependent intracellular signals that promote the release from the cell surface of pro-HB-EGF through the matrix metalloproteinase-mediated proteolytic cleavage of this and other EGF-like growth factor precursors. Free, active HB-EGF binds to EGFR, leading to transactivation of the EGFR.
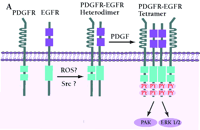
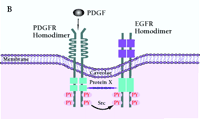
Mechanism of PDGF-induced EGFR transactivation. A. Model of basal PDGFβR-EGFR heterodimer formation and signal transduction. As proposed by Saito et al., PDGFβR-EGFR heterodimers exist in unstimulated cells (11). The formation of the heterodimer provides a scaffold for other molecules that induces EGFR transactivation and downstream signaling. The constitutive interaction between PDGFβR and EGFR is regulated by ROS and Src family kinases. B. Alternative mechanism of EGFR transactivation in caveolae. Caveolae are the principal location of PDGFβR (14). Upon PDGF binding to PDGFβR, EGFR is activated by Src-mediated phosphorylation prior to the formation of EGFR homodimers. The heterodimers between EGFR and PDGFβR may bind each other indirectly as a part of a large cross-linked complex involving unknown proteins.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

H. Shelton Earp III, MD, is a Professor of Medicine and Pharmacology at the University of North Carolina at Chapel Hill, and is a Professor and Director of the Lineberger Comprehensive Cancer Center. Address correspondence to HSE. E-mail HSE{at}med.unc.edu.

Jun Han, PhD, is a Postdoctoral Investigator of Pharmacology at the University of North Carolina at Chapel Hill.

Lee M. Graves, PhD, is an Associate Professor of Pharmacology at the University of North Carolina at Chapel Hill.

