Act Locally: New Ways of Regulating Voltage-Gated Ion Channels
In recent years, the modulation of ion channels by phosphorylation has been a major focus of research, and practically every category of channel possesses putative phosphorylation sites. Indeed, most channels are directly modified by kinases and phosphatases. Recent studies of the interactions of non-opioid sigma receptors with voltage-dependent ion channels suggest yet another mechanism of signal transduction that appears to be independent of phosphorylation. Sigma receptors were first characterized as opiate receptors based on their binding to the agonist SKF10047 (1). However, this designation was abandoned when it was discovered that most other opiate receptor agonists and antagonists, including endogenous opiates, do not bind sigma receptors. In fact, characterizing sigma receptor binding became increasingly problematic, as pharmacological agents of very diverse chemical structure and receptor-selectivity were found to interact with sigma sites. To date, no endogenous sigma receptor ligands have been identified, although several categories of endogenous molecules can alter binding of canonical sigma receptor agonists. Candidate endogenous ligands include dopamine, serotonin, numerous steroids, and several peptides (most prominently neuropeptide Y)—their diversity further revealing the idiosyncratic binding properties of sigma receptors.
Indeed, the profligate binding of sigma receptors has made it difficult to ascribe functions to the receptor. Attributions range from higher-level cognitive processing to regulation of digestive, reproductive, and liver function. Nonetheless, sigma receptors are expressed in many tissues and probably play roles in several vital functions. Recently, several papers by Jackson and colleagues have elucidated a specific cellular function for sigma receptors in neuroendocrine cells of the pituitary gland. These findings not only offer to tie together the disparate proposed functions but, more interestingly, indicate a novel mechanism of cellular signal transduction. Originally, Wilke et al., using whole-terminal patch clamp in posterior pituitary slices, showed that the dopaminergic agonists (±)-2-(N-phenethyl-N-propyl)amino-5-hydroxytetralin (PPHT) and U101958 reduced two neurohypophysial potassium currents (2). Both drugs also bind to sigma receptors, and further studies with receptor-specific agonists and transgenic mice eliminated any role for dopamine in the effect on the pituitary currents. Additional work by the same group demonstrated that several other, non-dopaminergic, sigma receptor drugs (i.e., SKF10047, pentazocine, ditolylguanidine, haloperidol, and apomorphine) also reduced the neurohypophysial potassium currents (3). This specific and tractable cellular assay was then used to identify cellular signaling pathways that mediate sigma receptor effects. Evidence from earlier studies suggested that altering the function of G proteins could alter the binding of sigma ligands, although no unambiguous functional coupling between G proteins and sigma receptors was ever established (4). Using activators and inhibitors of G proteins as well as the non-hydrolyzable ATP analog AMPPcP, Lupardus et al. found the quite surprising and unexpected result that the sigma receptor–induced reduction of neurohypophysial potassium currents was independent of G protein activity and phosphorylation (5).
These studies suggest that the binding of sigma receptors to extracellular ligands could create a non-traditional cellular signal that alters the functions of ion channels. That this effect occurs locally in the membrane was demonstrated by comparing the efficacy of sigma agonists on channels in different patch conformations. For example, bath-applied SKF10047 reduced the current in channels from excised patches in the “outside-out” conformation, where bath-applied ligand is available directly to the extracellular binding domain of the receptor but cytoplasmic signaling intermediates are not available to the intracellular domains of the receptor. This occurred even when AMPPcP was included in the pipette to eliminate the possibility that small amounts of cytoplasm taken up during patch excision could mediate the effect. Furthermore, when channels in a small patch of membrane were isolated from the bath but still in contact with cytoplasmic signaling molecules (the cell-attached patch conformation), bath-applied sigma agonists had no effect on the channels in the patch. Thus, the receptor and the channel are in close proximity and do not require any soluble factors for signal transmission (5). In oocytes, heterologously expressed sigma receptors immunoprecipitate with the voltage-dependent potassium channels Kv1.4 and Kv1.5 (6), further suggesting that sigma receptors and potassium channels interact directly or through closely associated intermediates. Like the two pituitary potassium currents, both channels also show reductions in peak current amplitude when coexpressed with sigma receptors and treated with sigma agonists. Furthermore, GFP-labeled sigma receptors are expressed at the cell surface where they can interact directly with coexpressed potassium channels. Thus, in response to extracellular signals, sigma receptors are capable of modulating channel function by interacting either with channel proteins or some very closely associated membrane constituent.
The most interesting unresolved questions from the new results on sigma receptors are concerned with the mechanism of signal transduction: How do sigma receptors act on ion channels to reduce current? The observations to date present several constraints. In addition to the lack of involvement of G proteins and kinases or phosphatases, the interaction occurs either within the membrane or among components that are closely associated with the inner surface of the membrane and does not include any diffusible second messengers. In spite of the close spatial requirement, it still is not clear whether a direct interaction exists between the sigma receptors and the ion channels. Given these constraints, receptor activation from the unliganded, resting state (Figure 1A⇓) could result in a steric alteration of the receptor molecule that either allows or alleviates a specific interaction with a channel or a closely associated intermediate (Figure 1B and 1C⇓). Intermediates could include ion channel β subunits or other components of the membrane such as lipids or cholesterol.
Another intriguing possibility arises from the amino acid sequence of sigma receptors (7). The sigma-1 receptor, which is the isoform used in the studies of potassium channel regulation described above, shares some sequence similarity with sterol isomerases, particularly the yeast ergosterol biosynthetic enzyme ERG2 (8, 9). Sterol isomerases catalyze the formation of cholesterol suggesting that cholesterol-rich membrane microdomains, such as lipid rafts, may be altered by activated sigma receptors. Although sigma receptors fail to rescue yeast ERG2 lethal mutants (8), this might be explained by differences in yeast and mammalian sterol substrates, and cannot be used to definitively rule out sigma-mediated sterol isomerization in mammalian cells. It will be of considerable interest to determine if sigma receptors possess endogenous sterol isomerase activity with mammalian substrates. Interestingly, Kv1.5, one of the channels shown by Aydar et al. to be a target of sigma receptors (6), is also associated with cholesterol-rich lipid rafts, and cholesterol depletion dramatically alters kinetic properties of Kv1.5 (10, 11). Even if sigma receptors no longer retain isomerase activity, they may still retain the capacity to bind to cholesterol (Figure 1D⇓) or its precursor, and thereby alter the organization of lipid domains within the plasma membrane as has been suggested in other raft-associated signaling systems (12).
Sigma receptor modulation of potassium channels presents cellular biologists with a novel mechanism of signal transduction that is reminiscent of, but not identical to, the gating of the cardiac acetylcholine-regulated inward rectifier. Subsequent to ligand binding at muscarinic sites and the dissociation of Gi, potassium channels in heart cells are altered by a direct interaction with the βγ subunits (rather than with the GTPase-containing α subunit) (13). Indeed, the mechanism proposed (Figure 1B⇓) may reflect a similar type of protein–protein interaction between membrane-associate regions of sigma receptors and potassium channels as that observed to occur between βγ subunits and the delayed rectifier. The difference is that unlike G proteins, sigma receptors are not intermediates. Instead, the sigma receptor makes its own direct link from the extracellular ligand-binding event to a change in activity of a cellular target.
Are there other examples of the sigma type of signal transduction to be found? A number of proteins can interact with ion channels and possess extracellular domains of unknown function. These include the β subunit of the Slo Ca2+-dependent K+ channel (14), and the small transmembrane protein minK which acts a subunit for some voltage-dependent potassium channels (15). Although neither of these examples are known to possess extracellular binding partners, the new finding on sigma receptors has refined the theoretical framework for elucidating their mechanisms of action. More broadly, the sigma receptor form of signal transduction suggests that cells also have unique ways of responding within local microdomains. For example, although activation of traditional signaling pathways (i.e., utilizing kinases and phosphatases), can invoke global cellular activation such as the recruitment of new gene expression, it is undoubtedly beneficial for cells to respond to locally available cues only in the region of the cell exposed to the altered environment. This is especially relevant for large, complex cells that extend cellular domains over vast areas (e.g., neurons, glia, dendritic cells, fibroblasts) and for migratory developing cells that must respond to local guidance cues. Thus, it will be important to determine whether the functions of sigma receptors extend to other membrane proteins other than ion channels, and it will be of even more significance to find other local signal transducers.
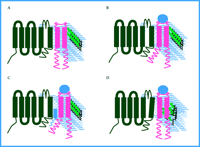
Putative models of sigma receptor modulation of ion channels. The plasma membrane is diagrammed to contain an idealized voltage-dependent ion channel (black) in close proximity to a sigma receptor (pink). The phopsholipid bilayer is depicted in blue and cholesterol molecules are green. When the unbound receptor and ion channel (as shown in A) binds to a ligand (blue circle), a conformational change occurs in the transmembrane or intracellular domains (or both) of the sigma receptor so that channel accessibility is either directly (as in B) or indirectly (as in C) regulated. Alternatively, ligand binding may result in local changes of cholesterol structure due to the activity of, or binding to, sterol isomerase-like sequences in the sigma receptors (D).
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Sharen E. McKay, PhD, is an Associate Research Scientist in the Pharmacology Department at Yale University School of Medicine and an Adjunct Professor of Psychology at Manhattanville College.

Leonard K. Kaczmarek, PhD, is a Professor of Pharmacology and a Professor of Cellular and Molecular Physiology at Yale University School of Medicine. Address correspondence to LK. E-mail leonard.kaczmarek{at}yale.edu.

