Fibrin Mechanisms and Functions in Nervous System Pathology
Abstract
In brain physiology, cerebrovascular interactions regulate both, vascular functions, such as blood vessel branching and endothelial cell homeostasis, as well as neuronal functions, such as local synaptic activity and adult neurogenesis. In brain pathology, including stroke, HIV encephalitis, Alzheimer Disease, multiple sclerosis, bacterial meningitis, and glioblastomas, rupture of the vasculature allows the entry of blood proteins into the brain with subsequent edema formation and neuronal damage. Fibrin is a blood-derived protein that is not produced by cells of the nervous system, but accumulates only after disease associated with vasculature rupture. This review presents evidence from human disease and animal models that highlight the role of fibrin in nervous system pathology. Our review presents novel experimental data that extend the role of fibrin, from that of a blood-clotting protein in cerebrovascular pathologies, to a component of the perivascular extracellular matrix that regulates inflammatory and regenerative cellular responses in neurodegenerative diseases.
Neurovascular Dynamics in Health and Disease
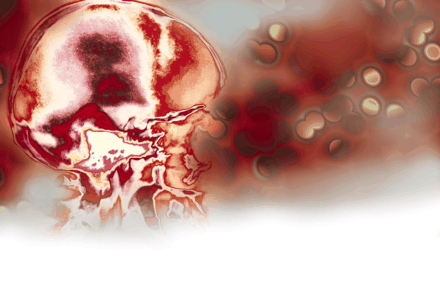
Neurons and astrocytes are in close proximity to brain endothelial cells and regulate vascular homeostaasis and cerebral microcirculation. Disruptions to this homeostatic balance contribute to several nervous system pathologies including ischemic events, such as stroke, as well as neurodegenerative diseases, such as multiple sclerosis (MS) and Alzheimer Disease. The role of the vasculature in the nervous system has traditionally been considered one in which the blood supplies nutrients to and removes toxic products from the brain (1). Recent evidence has expanded the role of the vasculature from that of nutrition delivery and removal to a role as a major regulator of nervous system pathophysiology. For example, experiments using genetic methods to eliminate nerves have demonstrated a common developmental mechanism for nerve and blood vessel branching (2), suggesting that blood vessels might provide guidance cues for sympathetic axons en route to their intervention targets (Figure 1A⇓). In addition to neurons, in the developing retina, astrocyte distribution colocalizes exquisitely with the developing microvasculature (3). The identification of artemin as a vascular-derived neurotrophic factor that acts as a guidance protein for the nerves to reach their final targets has provided a molecular understanding of the interactions of the vasculature with the nervous system (4). Furthermore, integrins are involved in the maintainance of blood vessel integrity, because mice deficient for av integrin develop cerebral hemorrhages (5). In addition to development, similar mechanisms drive adult neurogenesis. The provision of an “angiogenic niche” for local neurogenesis has been reported in the dentate gyrus of the hippocampus (6), and vascular endothelial growth factor (VEGF) is a major player in neuronal recruitment in the adult ventricular zone (7). In addition, astrocytic calcium signaling at the gliovascular interface has been implicated in functions such as blood-flow circulation, metabolic trafficking, and water homeostasis (8) (Figure 1B⇓). Electrophysiological and hemodynamic imaging approaches also have shed light on the coupling of neuronal activity and vascular responses (9). For example, these studies revealed the relationship between neurovasculature-controlled oxygen metabolism and regulation of local synaptic activity (9). The vasculature plays an important role in the visual system: the retinal blood vessels lead to rewiring of the eyes’ cortical projections, imprinting an image of the retinal vascular tree onto the primary visual cortex (10). Overall, nervous system cells, such as perivascular neurons and astrocytes, cross-talk with vascular endothelial cells and form a functional unit, defined as the neurovascular unit (11) (Figure 2A⇓). The neurovascular and gliovascular interfaces determine a variety of physiological functions of the nervous system including neuronal development, synaptic activity, metabolic trafficking, and adult neurogenesis.

The neurovascular and gliovascular interface in nervous system physiology. A. Arteries are specifically aligned with peripheral nerves in forelimb skin. B. End feet of astrocytes surrounding blood vessels. Reprinted from (2); Cell, 109, Mukouyama et al. “Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the ski,” 693–705, (2002), with permission from Elsevier; and from (8); J. Neurosci., 23, Simard et al. “Signaling at the gliovascular interface,” 9254–9262, (2003) by the Society for Neuroscience.
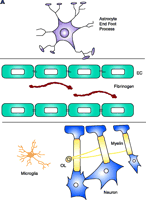
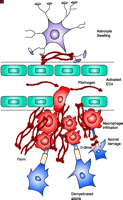
Schematic representation of fibrin extravasation in the neurovascular unit. A. Neurons and astrocytes are in close proximity with brain endothelial cells and their interactions form a functional unit, termed the neurovascular unit (11). Tight junctions between the endothelial cells (EC) confine fibrinogen in the vasculature. B. After vascular damage leading to disruption of the BBB, fibrinogen gains access within the CNS parenchyma with consequent fibrin formation. In MS, fibrin deposition correlates with the sharp border of demyelination (141), colocalizes with macrophage infiltration (80), with axonal damage (81, 92), and with astrocyte swelling and tight junction abnormalities (82). Fibrinogen molecule adapted from (33).
Although there is a causal interaction between the nervous system and vascular homeostasis, a physical and metabolic barrier between the brain and the systemic circulation––namely, the Blood–Brain Barrier (BBB)––regulates the entry of blood proteins from the vasculature into the nervous tissue (12). The cellular and molecular basis of the BBB has been reviewed elsewhere (13). Disruption of the BBB is a hallmark of many central nervous system (CNS) pathologies, including stroke (14), HIV encephalitis (15), Alzheimer Disease (16, 17), MS (18), spinal cord injury (19), brain glioblastoma (20, 21), and bacterial meningitis (22). In these diseases, components of the blood leak inside the brain with subsequent hemorrhage, edema formation, and neuronal damage occurring.
The amount of fibrinogen, a protein normally present in the blood at concentrations ranging from ~1.5–4 mg/ml, is known to increase in pathological conditions (23). The role of fibrinogen is well established in blood clotting and circulation. In conditions of BBB disruption, fibrinogen leaves the vasculature (i.e., extravasates) and enters the brain parenchyma where it alters the extracellular matrix (ECM) composition of the neurovascular unit (Figure 2B⇑). Injury responses induce local procoagulant activity (24), resulting in conversion of fibrinogen to fibrin by the action of perivascular tissue factor (25). The procoagulant proteins necessary for fibrin formation are present in the brain parenchyma; the concentrations of these proteins increase after injury (26). Inflammatory mediators, such as tumor necrosis factor–α (TNFα) and endotoxin, in addition to their proinflammatory activities, may also contribute to fibrin formation by increasing the expression of key molecules of the coagulation cascade (27). The scope of this review is to present the potential mechanisms that fibrin utilizes to affect regenerative and disease mechanisms during neurologic disease.
Fibrinogen
Fibrinogen is a soluble 340-kDa dimeric glycoprotein synthesized by the liver. Each monomeric subunit is composed of three nonidentical polypeptide chains, designated Aα, Bβ, and γ that are linked together by disulfide bonds. Like other blood proteins, fibrinogen is secreted into the plasma and circulates throughout the body. Fibrinogen is most classically known for its role in blood coagulation. Upon initiation of the coagulation cascade, the serine protease thrombin cleaves peptides from the N-terminal regions of the Aα and Bβ chains. The cleavage of these peptides, termed fibrinopeptide A and fibrinopeptide B, exposes polymerization sites and initiates the formation of a polymer consisting of noncovalently linked protofibrils, which, upon binding of activated coagulation factor XIII, form an insoluble fibrin clot [for review see (28)]. In addition, the released fibrinopeptide B can independently modulate inflammatory responses by acting as a chemoattractant for leukocytes (29). Recent studies on the crystal structure of fibrinogen and fibrin (30–32) have led to the development of a model for fibrin formation (33) and have suggested a structural basis for the diverse biological activities of fibrinogen and fibrin.
Fibrin is considered a provisional matrix because it is subjected to proteolytic degradation by plasmin. Plasmin, derived from the inactive zymogen plasminogen, degrades fibrin into soluble fibrin degradation products. Conversion of plasminogen to plasmin is regulated by two plasminogen activators (PA): the tissue type PA (tPA) and the urokinase type PA (uPA) (34). Plasmin and tPA both bind to fibrin and initiate its proteolytic degradation (28). Plasmin digestion of fibrin results in the production of two soluble fibrin degradation products, namely D-dimer and fragment E [for review see (35)]. D-dimer is extensively used in clinical practice as a marker for increased coagulation activity and risk predictors for thrombotic events (36). Experiments from cases of human disease or from mice genetically lacking components of the coagulation and proteolytic pathways have shown that fibrin exacerbates disease pathogenesis in several different tissues (37). Notably, genetic depletion of fibrinogen rescues the effects of plasminogen deficiency in liver, lung, and kidney damage, as well as in impaired wound healing, showing that fibrin is the major substrate for plasmin in vivo (38).
The ability of fibrinogen and its derivatives to act in different pathological settings can be attributed to fibrinogen’s ability to signal through receptors with widespread tissue expression and fibrinogen’s ability to bind a diverse group of proteins including growth factors, bacterial proteins, and other proteins that regulate coagulation and fibrinolysis. Within the context of this review, fibrin(ogen) receptors are defined as membrane-bound proteins that can transduce intracellular signals upon fibrin(ogen) binding (Table 1⇓), whereas fibrin(ogen) binding proteins are either soluble or anchored molecules that bind fibrin(ogen) but have no documented ability to directly transduce intracellular signals upon fibrin(ogen) binding (Table 2⇓). The functional consequences of these protein–fibrin(ogen) interactions range from blood coagulation and initiation of angiogenesis, to inflammation and propagation of infection.
Fibrin(ogen) Receptors, Signaling Pathways Induced, and Biological Functions
Fibrin(ogen) Binding Proteins and Biologial Functions
Fibrin(ogen) Receptors
The best documented fibrin(ogen)–receptor interactions are those with the integrin receptors. Fibrin(ogen) binds several families of integrins, including β1, β2, and β3 subtypes. Fibrin(ogen)–integrin interactions mediate a variety of cellular responses, including clotting and inflammation. Binding of the fibrinogen dimer to αIIbβ3 integrin on platelets causes adhesion and subsequent platelet aggregation necessary for blood coagulation (39). Fibrin(ogen) participates in inflammatory responses through at least three integrin types: α5β1, αMβ2 (also known as Mac-1 or CD11b/CD18), and αvβ3. These receptors are expressed on leukocytes, macrophages, and monocytes (40), and contribute to inflammatory responses. Fibrin(ogen) signaling through αMβ2 can elicit different responses, depending upon the cell type and intracellular signaling pathways involved. Signaling via the mitogen-activated protein kinase (MAPK) or the phosphoinositide-3 kinase (PI3K) pathway elicits neutrophil survival (41), whereas stimulation of the NF-κ B pathway causes increased of cytokine gene expression in monocytes (42). Fibrin(ogen)–integrin interactions reveal a potent role for fibrinogen in inflammation [for review, see (26)].
Other receptors besides the integrins bind fibrin(ogen) and elicit cellular effects, including intracellular adhesion molecule-1 (ICAM-1) and vascular endothelial cadherin (VE-cadherin). Binding of fibrin(ogen) to ICAM-1 or to VE-cadherin also promotes cell adhesion (Table 1⇑). ICAM-1 is additionally involved in fibrinogen-mediated migration of neutrophils, which occurs via a Rho-GTPase intracellular signaling pathway (43, 44). Endothelial cell survival is also mediated by binding of fibrinogen to ICAM-1 via the MAPK pathway (45). VE-cadherin interacts with the N-terminal fragment of fibrin to elicit endothelial cell migration and capillary tube formation (46). The elucidation of the signaling mechanisms involved in this process will be important for furthering our understanding of the role that fibrin(ogen) plays in vascular homeostasis and angiogenesis.
Fibrin(ogen) Binding Proteins
The classical view of fibrin(ogen) as a matrix protein has been expanded upon within recent years: Not only does fibrin(ogen) serve as an anchoring matrix for cell attachment, but it also serves as a molecular “net” to capture growth factors, thereby elevating the local concentration and potency of growth factors. Through interactions with various binding proteins, fibrin(ogen) participates both in beneficial physiological processes (e.g., coagulation) and in pathogenesis (Table 2⇑).
The binding interactions between ECM proteins (e.g., fibronectin) and fibrin(ogen) regulate adhesion and migration of fibroblasts during wound healing (47, 48). Gelsolin, an actin-binding plasma protein, binds to fibrin(ogen) and becomes incorporated into clots (49). Binding of fibrin(ogen) to insulin-like growth factor binding protein-3 (IGFBP-3) may be involved in wound healing. IGFPB-3 binds insulin-like growth factor-1 (IGF-1), a multipotent protein with mitogenic and chemotactic properties that can induce stromal-cell migration necessary for wound repair (50). Interactions of fibrin(ogen) with other binding proteins may play a role in tissue repair and angiogenesis through effects on endothelial cells. Basic fibroblast growth factor (bFGF, FGF-2) can bind to fibrin or to fibrinogen to stimulate the proliferation of endothelial cells (51, 52). Francis and colleagues have also shown that VEGF can bind fibrinogen and fibrin and support endothelial cell proliferation (53). In addition, upon platelet activation, fibrin formation is accompanied by translocation of protein kinase A (PKA), vitronectin, and plasminogen activator inhibitor-1 (PAI-1) onto the fibrin fibers, a mechanism proposed to phosphorylate vitronectin and initiate fibrinolysis (54).
Fibrin(ogen) may also serve a role in microbial pathogenesis. Common bacteria such as Staphylococcus and Streptococcus express proteins that can bind to fibrin(ogen). Staphylococcus epidermidis and Staphylococcus aureus express cell wall–anchored adhesin proteins that bind fibrin(ogen), promoting bacterial adhesion and contributing to anticoagulant properties by masking fibrin’s binding sites (Table 1⇑) (55). S. aureus also secretes soluble fibrin(ogen)-binding proteins that prevent coagulation (55). Group A streptococci express cell wall–anchored proteins M1 and M5, which bind fibrin(ogen) with high affinity and are thought to mediate the bacteria’s survival (56).
Fibrin(ogen) in the Nervous System
The nervous system, especially under inflammatory conditions, provides molecules related to fibrin––such as proteins of the coagulation and fibrinolytic cascade––as well as fibrin receptors, and intracellular signaling molecules activated by fibrin [reviewed in (26)]. Recent studies have identified Schwann cells in the peripheral nervous system (PNS) and macrophages in the CNS as two major cell types affected by fibrinogen extravasation. We present experimental data together with evidence from human disease to uncover the fibrin-dependent mechanisms that inhibit regeneration in the peripheral nervous system and exacerbate inflammatory demyelination in the central nervous system.
Fibrin in Peripheral Nerve Regeneration
Failure of nerve regeneration does not arise from an inability of central neurons to elongate and remyelinate, but rather arises from the nonpermissive nature of the CNS environment (57). ECM proteins play a major role in peripheral nerve development and regeneration (58) and affect biological properties of neurons and Schwann cells (the myelinating cells of the PNS). Many of the signals that trigger Schwann cell proliferation and myelin synthesis are derived from the molecules in the ECM (59, 60), including laminin (61, 62) and laminin β1 integrin receptors (63, 64). Leakage of fibrinogen from blood vessels can alter the composition of the ECM after nerve injury. In conditions of nerve injury the local vasculature is damaged concomitantly with the nerve, resulting in leakage of fibrinogen from surrounding blood vessels, resulting in fibrin deposition (65). Immunohistochemical analyses of sciatic nerves reveal fibrin deposits that correlate spatially and temporally with nerve degeneration, whereas fibrin clearance correlates with nerve regeneration (65, 66).
Fibrin clearance (i.e., removal) primarily results from the activity of plasmin. Amounts of plasminogen activators are increased in response to experimental nerve injury (67). For example, tPA levels are increased following sciatic nerve injury, and increased proteolytic activity after injury is primarily dependent on plasminogen activation (65). The upregulation of tPA has a functional role in the nerve, however, because axonal degeneration is exacerbated in mice lacking either tPA or plasminogen (65). Behavioral studies also demonstrated delayed functional recovery after injury in mice lacking tPA (68), further supporting the initial histopathological observations that lack of tPA exacerbates nerve damage. Genetic or pharmacologic elimination of fibrinogen rescued tPA-deficient or plasminogen-deficient mice from exacerbated axonal damage, thus implicating tPA-mediated fibrinolysis as a protective mechanism in conditions of nerve injury (65). Overall these data––when combined with the temporal regulation of fibrin deposition in axonal degeneration, and the correlation of fibrin clearance with axonal regeneration––suggest a role for fibrin clearance in nerve repair.
The role of fibrin in neuronal regeneration was studied in a model of sciatic nerve crush injury. Mice depleted pharmacologically of fibrin [through the use of the snake venom ancrod (Figure 3⇓)] or genetically deficient for fibrin(ogen) (69) exhibited an earlier onset of nerve regeneration after injury, as compared to nerve regeneration in wild-type control mice (66). Functional nerve regeneration after injury is a multistep process that requires remyelination and regrowth of the damaged axons to their targets. Fibrin primarily affects nerve regeneration at the level of myelination: Transcription of myelin genes is accelerated in fibrindeficient mice after nerve injury (66). Because the major effect of fibrin is on the myelination process, Schwann cells were utilized to determine the cellular and molecular mechanisms of fibrin action in the PNS. Fibrin induced Schwann cell proliferation via phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) in vitro. In addition, fibrin appears to regulate the expression of fibronectin and p75 Neurotrophin receptor (p75NTR) in Schwann cells (66) (Figure 4⇓).
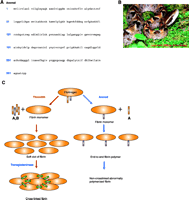
Fibrinogen depletion by the snake venom protein ancrod. A. Ancrod is a 258-amino acid serine protease derived from the Malayan pit viper, Calloselasma (Agkistrodon) rhodostoma (photo provided by Mardi Snipes) (B), with thrombin-like specificity for fibrinogen (142, 143). C. Ancrod cleaves only the fibrinopeptide A without removing fibrinopeptide B, thus resulting in improper fibrin polymer formation. Ancrod-cleaved fibrinogen cannot be crosslinked by Factor XIII (transglutaminase) to form fibrin and, thus, is subsequently degraded and depleted from the circulating blood supply.
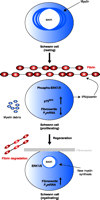
Proposed mechanism for the effects of fibrin in peripheral nerve remyelination. Myelinating Schwann cells in the adult normal sciatic nerve have established axonal contacts and produce myelin that enwraps the axons. After injury, axons degenerate, myelin is degraded, and fibrin is deposited. Fibrin deposition regulates Schwann cell numbers by increasing the phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) and by increasing the protein expression of p75 neurotrophin receptor (p75NTR). The combined action of the proliferating ERK1/2 pathway and the p75NTR pathway sustains the Schwann cells in a nonmyelinating state, as evidenced by the shut down of myelin genes transcription [e.g., the myelin protein zero (P0) gene], and decreased amounts of fibronectin transcripts. Downregulation of the P0 or the fibronectin gene could either be due to the increased ERK1/2 phosphorylation as observed in transformed cells (144), and/or by another mechanism. When fibrin is degraded during nerve regeneration, ERK1/2 becomes unphosphorylated, p75NTR levels decrease, fibronectin is produced and deposited in the nerve, and Schwann cells transcribe myelin genes and remyelination is initiated. Up-regulation of P0 associated with downregulation of p75NTR is a characteristic change associated with myelination (145). Reprinted from (66); Neuron, 14, Akassoglou et al. “Fibrin inhibits peripheral nerve remyelination by regulating Schwann cell differentiation,” 861–875, (2002), with permission from Elsevier.
Cell migration is an additional function in Schwann cells affected by fibrin (70). Following nerve injury, portions of the axon distal to the damaged site degenerate and are cleared by macrophages. Schwann cells then migrate through the ECM in the proximity of the newly formed axons. Subsequently, the Schwann cells proliferate, and differentiate into a myelinating state (71, 72). Schwann cells migrate through ECM proteins such as fibronectin and laminin (72); however, after sciatic nerve crush, fibrin incorporates into the ECM and colocalizes to the nerve distal to the crush site along with fibronectin (66, 70). Schwann cells cannot migrate on fibrin matrices, thus their migration is impaired, in a dose-dependent manner, on mixed fibrin–fibronectin matrices (70). Schwann cell migration on fibronectin is mediated by an Arg-Gly-Asp (RGD)-dependent integrin (73). Fibrin might mediate its inhibitory effects by antagonizing integrin signaling in Schwann cells. Although this hypothesis has yet to be proven, αVβ8 integrin (an RGD-sensitive integrin) does act as a fibrin receptor on Schwann cells (74). Fibrin binding to αVβ8 integrin might provide a mechanism by which fibrin inhibits Schwann cell myelination, migration, and nerve regeneration. Thus, the infiltration of fibrin into the ECM following nerve injury can delay the innate regenerative capacity of the peripheral nerve. Consequently, the clearance of fibrin from damaged neurons is a requirement for optimal regeneration. A comprehensive understanding of the regulation of coagulation and fibrinolysis in the nervous system promises to provide new targets for the pharmacologic enhancement of nerve regeneration.
Fibrin in Multiple Sclerosis
MS is a chronic inflammatory demyelinating disease of the nervous system in which an inflammatory process is associated primarily with destruction of myelin sheaths and later, with axonal damage leading in permanent functional deficits, such as paralysis, and loss of vision (75). Vascular disruption is one of the earliest events associated with the initiation of MS pathology (Figure 5A⇓) (76).
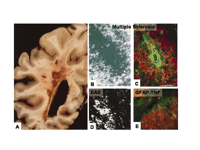
Fibrin deposition in inflammatory demyelinating plaques of MS and respective animal models. A. Unstained gross coronal section of brain shows typical, slightly hemorrhagic, periventricular, and parenchymatous lesions. MS plaque viewed under polarized light (B) and double labeled for fibrinogen (green) and macrophages (red) (C). High macrophage density is associated with fibrinogen leakage. Two rodent models for MS also reveal fibrin depositions in inflammatory demyelination. D. In EAE, foci of fibrin deposits are observed perivascularly. E. In a TNF-transgenic mouse model for MS, fibrin deposition (red) is present in regions of macrophage infiltration (green). From (80); Brain 120, Gay et al., “The application of multifactorial cluster analysis in the staging of plaques in early multiple sclerosis. Identification and characterization of the primary demyelinating lesion,” 1461–1483 (1997), by permission of Oxford University Press. From (98); Fed. Proc. 35, Paterson, “Experimental allergic encephalomyelitis: Role of fibrin deposition in immunopathogenesis of inflammation in rats,” 2428–2434 (1976), by permission from FASEB. From (146); Poser, An Atlas of Multiple Sclerosis. The Parthenon Publishing Group Inc, New York, NY, p. 60 (1998), by permission of CRC Press.
In 1863, Eduard Rindfleisch, a German pathologist who analyzed post-mortem brain samples from MS patients, made the observation that a blood vessel was present at the center of each lesion. He wrote: “If one looks carefully at freshly altered parts of the white matter...one perceives already with the naked eye a red point or line in the middle of each individual focus,...the lumen of a small vessel engorged with blood....All this leads us to search for the primary cause of the disease in an alteration of individual vessels and their ramifications; All vessels running inside the foci, but also those which traverse the immediately surrounding but still intact parenchyma are in a state characteristic of chronic inflammation” (77). Modern diagnostic techniques for MS, such as Magnetic Resonance Imaging (MRI), monitor the uptake of intravenous administered contrast material in new lesions of the brain. MRI monitoring of BBB breakdown in MS patients has been exploited for diagnosis, progression, observation of new lesion formation, and clinical drug trials for MS. Importantly, this disturbance to the BBB is an early event in the development of MS and is thus, believed to precede symptoms and new lesion formation (76). As such, evidence for BBB abnormality, including vascular changes and the presence of serum proteins within the demyelinated plaques, has been examined in several studies using immunohistochemical stainings on MS plaques.
Vascular abnormalities associated with MS include an accumulation of perivascular fibrin, which is suggestive of increased microvascular permeability of the BBB (78). Examination of chronic inactive, silent lesions using ultrastructural techniques reveals extensive fibrin deposition around small blood vessels, further indicating increased BBB disruption and leakage of blood proteins into the perivascular space (78). In addition, one of the earliest events coupled to the microvascular injury observed in acute MS lesions is focal endothelial cell activation associated with fibrin deposition (79). Fibrin deposits colocalize to inflamed blood vessels in conjunction with increased immune cell infiltration of the area (79). Similarly, fibrinogen deposition colocalizes with infiltrating macrophages within demyelinated plaques (Figure 5B and C⇑) (80), as well as with damaged axons stained for nonphosphorylated neurofilaments, a marker for axonal damage (81). Additional examination of vascular abnormalities in MS have shown a physical association of extravasated fibrinogen with astrocytic processes and altered endothelial cell tight-junction formation (82). Overall, these studies suggest that fibrin may be a contributing factor both for inflammation, glial scarring, BBB permeability, and axonal damage observed during MS pathogenesis.
Impaired Fibrinolysis in MS Lesions
The role of plasminogen activators, and in particular tPA, in other pathologic conditions of the nervous system (e.g., stroke, Alzheimer Disease, and excitotoxicity) has been recently reviewed elsewhere [for reviews (83–88)]. Components of the fibrinolytic pathway have also been examined from MS patients to analyze their contribution to disease pathogenesis. Analysis of cerebrospinal fluid (CSF) from MS patients revealed increases in tPA activity as compared to tPA levels in reference subjects (89, 90), and further examination of brain tissue from MS patients indicated increased tPA expression by neuronal cells (90, 91) and accumulation of fibrin D-dimer on demyelinated axons (92), suggesting that fibrin could be degraded in the MS plaques. However, as mentioned above, fibrin amasses in MS lesions; this may be explained by recent studies that show increased amounts of plasminogen activator inhibitors in MS lesions (81, 92). These studies revealed that tPA forms a complex with its inhibitor PAI-1, thereby preventing the degradation of fibrin. In agreement with these findings, PAI-1 has also been shown to increase in plasma (93) and CSF (90, 94) from MS patients as compared to PAI-1 levels in control subjects. Overall, these results highlight the impairment of the fibrinolytic system in the pathogenesis of MS.
Fibrin Depletion Protects from Inflammatory Demyelination
One of the best characterized models for inflammatory demyelination is experimental autoimmune encephalomyelitis (EAE), a model induced by immunization with proteins or peptides derived from components of the myelin sheath. Studies on EAE have provided insight in the understanding of the molecular and cellular mechanisms of inflammatory demyelination. As also observed in MS plaques, fibrin deposition is widespread in several animal species including guinea pigs (95), rats (96), and mice (97) upon induction of EAE. The role of fibrin in the development of EAE was initially studied nearly thirty years ago, when it was recognized that perivascular deposition of fibrin (Figure 5D⇑) correlated to paralytic episodes and that these two events rarely occurred independently (98). EAE-induced animals were then treated with ancrod (Figure 3⇑) to deplete their fibrinogen. Ancrod-treated animals exhibited decreased fibrin deposition, in agreement with reduced fibrinogen levels, as well as decreased paralysis. This was the first indication that fibrin depletion may be beneficial in ameliorating the consequences of inflammatory demyelination. Further studies have confirmed these observations, including the use of a separate defibrinating agent, batroxobin, to show reduced clinical scores in models of EAE (99).
Components of the fibrinolytic pathway have also been examined in EAE and parallel the results from MS patients showing both an increase in tPA as well as PAI-1 (100). The role of tPA has been additionally characterized during the course of EAE through the use of tPA-deficient mice which were subjected to myelin oligodendrocyte glycoprotein-induced EAE (101). These mice had a delayed initiation of the disease, but an impaired recovery phase, implying a complex role for tPA as detrimental in early periods of inflammatory demyelination but protective at later stages. Previous studies using proteolytic inhibitors have also shown the complex functions of proteases in EAE (102, 103). In addition, examination of fibrinolysis in spinal cords taken from rats subjected to cell-transferred EAE showed an abrupt decrease in fibrinolytic capacity occurring in conjunction with the onset of clinical signs of EAE (104–106) and increasing during recovery, without ever reaching physiological levels (107). These observations are in accordance with impaired fibrinolysis observed in MS plaques (92, 108).
Recent studies utilizing a transgenic mouse model for MS (109–111) by expressing TNFα in astrocytes (112) analyzed the role of fibrin in inflammatory demyelination. This model develops inflammatory demyelinating plaques that differ from those observed in EAE owing to the development of oligodendrocyte apoptosis and axonal damage (113). Recent studies in MS have underscored the involvement of oligodendrocyte apoptosis (114, 115) and axonal pathology (116) in MS pathogenesis, suggesting the importance of an animal model exhibiting those characteristics (117). In TNFα-transgenic mice, fibrin immunostaining revealed that fibrin deposition preceded inflammatory cell infiltration and formation of demyelinating plaques, and later correlated with the formation of demyelinating lesions [(118) and Figure 5E⇑]. In addition, demyelinated spinal cords showed impaired ability to stimulate proteolysis at sites of lesion, suggesting impairment in fibrin degradation. Crossing the TNF-transgenic mouse with a fibrinogen-knockout mouse (69) significantly prolonged their lifespan as compared to the lifespan of the parental TNF-transgenic mice, and delayed the onset of paralysis and inflammatory demyelination (118). Additionally, pharmacologic depletion of fibrinogen of the TNF-transgenic mice (through the use of ancrod) inhibited both the onset of inflammation and demyelination. Overall, the combination of mouse genetics and pharmacologic intervention has demonstrated the involvement of fibrin in the initiation of inflammatory demyelination and the benefits of fibrin depletion in models of MS with different disease etiology and pathologic manifestation. MS is a disease with profound heterogeneity in its clinical course, neuroradiological appearance of the lesions, and involvement of susceptibility gene loci. For these reasons, response to therapy is varied (119). The positive effect of prophylactic fibrin depletion in two models of inflammatory demyelination with diverse etiology and histopathologic characteristics [reviewed in (113)] indicate the potential for therapeutic benefits of anticoagulants that target fibrin in inflammatory demyelination.
Fibrin Induces Macrophage Activation
The mechanism by which fibrin mediates its effects in inflammatory demyelination is unknown, but one indication as to a potential cellular target comes from the observation that genetic depletion of fibrin reduces inflammation in a mouse model for MS (118). Fibrinogen is involved in inflammatory reactions where it regulates leukocyte–endothelial cell interactions (120) and mediates the release of proinflammatory cytokines (121, 122) and chemokines (123). In addition, use of the macrophage cell line RAW264.7 showed that immobilized fibrinogen could induce activation of macrophage cells (118). Changes in cell morphology were similar to those observed by administration of lipopolysaccharide (LPS), a potent activator of macrophages, and functionally resulted in decreased cell proliferation. Activated macrophage cells are known mediators of MS pathology and are critical to the initiation of inflammatory demyelination (124). Evidence that fibrin deposition precedes the development of clinical symptoms and demyelination in models for inflammatory demyelination (118, 125) and MS (79) implicates fibrin as a potential activator of infiltrating macrophage cells during the initiation of inflammatory demyelination. Further examination of the mechanisms that fibrin utilizes in nervous system pathogenesis will provide further insight into its role as an initial mediator of inflammation in MS.
Other Nervous System Diseases
Although most evidence on the role of intracerebral perivascular fibrin deposits in nervous system pathology comes from studies of MS lesions, the role of fibrin might extend to a variety of neurodegenerative diseases associated with vascular damage and BBB disruption. For example, in acute ischemic stroke there is significant disruption of the BBB associated with significant hemorrhagic transformation. Recently, the importance of cerebrovascular interactions for the development of stroke has been highlighted (11). Athough fibrinolytic therapies have been used extensively for the treatment of stroke patients, the role of extravasated fibrin on the rehabilitation of the CNS after the primary neuronal damage caused by stroke remains to be elucidated. In spinal cord injury there is extensive BBB disruption (19) and patients with spinal cord transection exhibit increased fibrinogen levels and fibrin degradation products (126). Additionally, axons fail to regenerate over long distances mainly due to the non-permissive nature of the CNS environment (127), suggesting that alterations of the extracellular matrix composition due to fibrin deposition might affect regenerative processes. In accordance, mice deficient for tPA show increased neuronal damage after spinal cord injury (128). Increased formation of fibrin D-dimer is associated with deep vein thrombosis (DVT) in spinal cord injury patients (129). Anticoagulants are currently used and several are in clinical trials for therapy of DVT and thromboembolism of spinal cord patients (130). In Alzheimer Disease, cerebrovascular dysfunction is a feature of neurodegeneration (131). Intracerebral hemorrhages are observed in a fraction of AD patients; a higher amount of BBB disruption is believed to occur in a focal and transient manner (132). This hypothesis is supported by the fact that amyloid β-peptide (Aβ), in addition to its role in neuronal degeneration, regulates endothelial cell damage (133). The relationship between fibrin(ogen) and AD has yet to be explored. Aβ strengthens fibrin, however, leading to denser, more compact clots (134). In addition, Aβ, similar to fibrin, can stimulate tPA-mediated plasmin formation; subsequent to this, Aβ is degraded by plasmin (135, 136). tPA-mediated Aβ degradation inhibits Aβ-induced neurodegeneration (136); therefore, it is possible that factors that regulate extracellular matrix composition and remodeling at the neurovascular interface can regulate disease pathogenesis.
Final Comments
Four unique levels of complexity characterize the role of the fibrinogen molecule in disease, including nervous system pathology. First, fibrinogen exists in several different molecular forms. It is present as a monomer, or dimer fibrinogen, a fibrin polymer, cross-linked fibrin, or as soluble fibrin degradation products. Derivatives of fibrinogen have diverse biological activities. Second, fibrinogen is composed of three chains with unique binding sites for different receptors. Third, fibrinogen regulates basic cellular functions, ranging from proliferation, adhesion and migration, to myelination, infection and cytokine expression, via a plethora of receptors and binding proteins with different temporal and spatial localization in cell types which are major players in pathophysiology. Fourth, fibrinogen is involved in a variety of physiological and pathological functions ranging from blood clotting and circulation, wound healing, muscle and nerve regeneration, atherosclerosis, glomerulonephritis, liver damage, and inflammatory demyelination.
Although the molecular mechanisms of fibrin action are not yet fully understood, depletion of fibrin is beneficial in a variety of pathological settings. Studies using animal models and experimental injuries in the nervous system suggest that fibrin could be a potential therapeutic target in nerve regeneration and inflammatory demyelinating diseases, such as MS. Although increased proteolytic activity is beneficial in nerve regeneration (65, 68), inflammatory demyelination (101), spinal cord injury (128), and remyelination (137), the exogenous administration of proteases, such as tPA, might not be the preferable therapeutic approach. tPA contributes to neuronal death in models of excitotoxic neuronal death (138) and cerebral ischemia (139). In addition, thrombolytic therapy with recombinant tissue plasminogen activator (rtPA), increases the risk of hemorrhagic transformation (140). Therapies aiming directly at inhibiting fibrin formation might be a safer approach. Further research to identify the cellular and molecular mechanisms that fibrin utilizes to mediate nervous system pathology will provide pharmacologic targets that will specifically block the actions of fibrin in nervous system disease.
Acknowledgments
We are grateful to Shoana Willis and Adrienne Andres for assistance with the preparation of this manuscript. This work was supported by The Wadsworth Foundation Young Investigator Award and the National Multiple Sclerosis Society research grant RG 3370 to KA.
- © American Society for Pharmacology and Experimental Theraputics 2004
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
-
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵

Katerina Akassoglou, PhD, is an Assistant Professor of Pharmacology at the School of Medicine at the University of California, San Diego. Her lab’s research interests focus on molecular and cellular mechanisms that are dictated by the extracellular environment after neurovascular damage and regulate degenerative and repair processes upon injury or disease in the nervous system. Address correspondence to KA. E-mail akass{at}ucsd.edu; fax 858-822-4027.

Ryan A. Adams, PhD, (second from left) is a postdoctoral fellow, and Melissa Passino, BA, (far left) and Ben Sachs, BA, (second from right) are Biomedical Sciences graduate students in the laboratory of Dr. Akassoglou in the Pharmacology Department at UCSD. Tal Nuriel, BS, (far right) is a research assistant at the Skirball Institute of NYU Medical Center at the lab of Dr. Moses V. Chao and a Visiting Scientist at the Department of Pharmacology at UCSD.

