RHO SIGNALING in Vascular Diseases
Abstract
Rho signaling pathways in vascular smooth muscle cells are highly activated in hypertension, a condition associated with a variety of vascular diseases, including restenosis injury and atherosclerosis. In this review we suggest that inflammatory cytokines and agonists of G protein–coupled receptors that activate Rho are effective triggers of vascular disease. Accordingly, Rho kinase inhibitors and statins may have therapeutic potential for preventing vascular disease characterized by Rho-mediated cell proliferation and gene expression.
Introduction
Appreciated as little more than a Ras homolog just a decade ago, the low-molecular-weight G protein Rho is now recognized as a formidable and versatile force in the signal transduction field. Rho signaling pathways, which likely serve homeostatic functions under normal physiological conditions, appear to be most highly activated under conditions of inflammation and injury. Whereas their recruitment may be of benefit for initiation of protective responses, their sustained activation may have pathological consequences in vascular smooth muscle. Activation of Rho in vascular smooth muscle is sufficient to produce vasoconstriction and thereby increase blood pressure; Rho also acts in concert with additional signaling pathways activated upon cell stress to produce changes in cell growth. This review will focus specifically on recent investigations of Rho signaling in vascular smooth muscle cell (VSMC) growth and a host of vascular pathophysiologies and occlusive diseases ranging from atherosclerosis to restenosis. A particular focus will be on findings that implicate increased Rho activity and signaling in a number of forms of experimental hypertension as well as in VSMC responsiveness to growth stimuli. This review will also detail the rapidly accumulating support for inhibitors of Rho signaling as important therapeutic treatments for atherosclerosis and restenosis injury.
Rho Activation and Regulation
G proteins bind GTP and thereby become activated as signal transducers; by virtue of their inherent GTPase activities, they act as elegant molecular switches, transducing a variety of extracellular signals in a regulated fashion. The Rho subfamily of small GTPases includes RhoA, RhoB, and RhoC; RhoA is the best characterized and has been shown to be involved in cell adhesion, migration and invasion (1, 2), cell growth responses (1, 3, 4) and gene expression (5, 6). Relatively less is known about RhoB and RhoC. Interestingly, RhoB has been shown to be immediately– early inducible (7) and to inhibit cell growth and induce apoptosis (8). RhoC, on the other hand, is expressed in certain forms of cancer, and its expression has been correlated with cell invasiveness (9–10).
Under basal conditions, Rho proteins are bound to guanine nucleotide dissociation inhibitors (GDIs) in the cytosol. Activation of small G proteins occurs through stimulation of guanine nucleotide exchange factors (GEFs), which promote the release of GDP in exchange for GTP. Once activated, Rho interacts with various effectors, including PRK1/2, mDia, rhotekin and the best-characterized effector, Rho kinase (also known as ROKα/ROCK-II and p160ROCK/ROCKβ). GTPase-activating proteins (GAPs) interact with Rho to terminate signal transduction.
Because active Rho translocates to the membrane, Rho activation was initially determined by Western blot analysis of membrane-associated protein; the binding of radiolabeled guanine nucleotides to Rho has also been examined. Currently, the most common method for measuring Rho activation is a “pull-down” assay in which GTP-bound Rho is affinity-precipitated using the Rho binding domain (RBD) of a Rho effector and subjected to Western gel analysis (11). A limitation of these assays is that they do not readily provide kinetic or spatial information; more recently, single-cell assays implementing immunostaining of Rho–RBD binding (12) or fluorescence resonance energy transfer (FRET)-based probes have thus become key to visualizing Rho activation and its relation to defined cellular events (13). Although the literature describing the activation of RhoA has become extensive, investigations into the activation of other Rho isoforms (RhoB, RhoC) remain limited.
A number of pharmacological tools have been employed to study Rho function. These include bacterial toxins such as the C3 exoenzyme from Claustridium botulinum, which ADP-ribosylates and thereby inactivates Rho proteins, and cytotoxic necrotizing factor 1, which activates Rho by deamidating a specific Glu residue (14–16). Statins, used clinically to reduce blood cholesterol levels (17), inhibit the isoprenylation, and therefore the membrane localization, of Rho (18, 19). Such inhibition may be one of the bases for the clinical effects of the statins. Dominant-negative mutants of both Rho and Rho kinase (20, 21) have been used to demonstrate their involvement in cellular functions, and pharmacological inhibitors of Rho kinase are also commonly used to establish Rho kinase–dependence of downstream responses.
Rho and Vascular Contraction
Vascular smooth muscle contraction is classically considered to be regulated by increases in intracellular Ca 2+ and activation of the Ca 2+ –calmodulin dependent myosin light chain kinase. This enzyme phosphorylates myosin light chain and thus promotes the cycling of actin–myosin interactions that generate contractile force. Another mechanism that contributes to the tonic component of vascular smooth muscle contraction, however, is Ca 2+ -sensitization, which occurs independent of increases in intracellular Ca 2+ and is mediated by RhoA. Among the earliest evidence for the involvement of G proteins in Ca 2+ -sensitization was the observation that GTP (or its stable analog GTγS) could elicit contraction of permeabilized vessels when constant Ca 2+ levels were maintained (22, 23). Subsequently, it was discovered that C3 exoenzyme, which ADP-ribosylates and inactivates Rho isoforms, blocks Ca 2+ -sensitization in response to norepinephrine plus GTP in per-meabilized vessels (24). The molecular mechanisms responsible for G protein–coupled receptor (GPCR)-stimulated, Rho-mediated Ca 2+ -sensitization involve the Rho effector Rho kinase. Rho kinase phosphorylates the myosin-binding subunit of myosin phosphatase 1 (MYPT1) and thereby renders it inactive (25). The accumulation of phosphorylated myosin light chain would thus increase contractility, a hypothesis corroborated by the finding that the catalytic subunit of Rho kinase produces contraction when added to permeabilized rabbit portal vein (26). Similarly, the Rho kinase inhibitor Y-27632 has been shown to block agonist and guanine nucleotide–stimulated contraction of rabbit mesenteric artery (27).
Rho and Vascular Smooth Muscle Cell Proliferation and Migration
Although a role for Rho in GPCR-stimulated vascular smooth muscle contraction is now well established, the involvement of Rho in GPCR-stimulated VSMC proliferation and migration has only recently come to the scientific forefront. Our studies using VSMC from the rat aorta have demonstrated that thrombin-stimulated DNA synthesis and migration are inhibited by exoenzyme C3 and Y-27632 (1). Urokinase-dependent VSMC proliferation also is dependent upon Rho (4). We have additionally observed that thrombin and angiotensin II (AngII) activate RhoA in rat aortic VSMC, whereas GPCR agonists such as norepinephrine and phenlyephrine, which do not act as mitogens, fail to activate RhoA (1). Intriguingly, although thrombin activates RhoA, and Rho is necessary for thrombin-stimulated DNA synthesis in rat aortic VSMC, adenoviral transfection of cells with activated RhoA is not sufficient to induce DNA synthesis. Rather, for mitogenic signaling in VSMC, it appears that RhoA collaborates synergistically with Ras, such that DNA synthesis is activated only by agonists that stimulate both pathways. This supports earlier studies indicating that activation of Rho is critical for Ras-induced cell cycle progression (28).
A number of downstream signals generated by activation of Rho might contribute to cell cycle progression and cell proliferation. One downstream effect of Rho is to reduce expression of cell cycle inhibitors such as the cyclin-dependent kinase inhibitors p21 Waf1/Cip1 and p27 Kip1 (28). Our studies have demonstrated that adenoviral expression of a constitutively active form of RhoA in rat aortic VSMC reduces p27 Kip1 protein levels. Direct activation of Rho with cytotoxic necrotizing factor-1 also decreases p27 Kip1 and increases DNA synthesis in human smooth muscle cells (26). Conversely, cellular levels of p27 Kip1 are increased when Rho function is inhibited by exoenzyme C3 or treatment with statins (29, 30).
An additional mechanism underlying the ability of Rho to positively influence cell cycle progression involves cytoskeletal effects on the nuclear translocation or phosphorylation of extracellular- regulated kinase (ERK). Cytoskeletal changes, in particular stress fiber formation and cell rounding, were the first responses reported to be affected by Rho proteins (31). Serotonin stimulation of bovine pulmonary artery VSMC leads to translocation of ERK to the nucleus, and this response is inhibited by treatment with either dominant negative RhoA or the Rho kinase inhibitor Y-27632 (32). We have similarly reported that mechanical stretch of neonatal rat ventricular cardiac myocytes leads to RhoA activation and nuclear translocation of phospho-ERK. Phospho-ERK translocation is inhibited by disruption of the actin cytoskeleton with jasplakinolide or disruption of caveolae with methyl-β-cyclodextrin (33). Although some reports suggest that it is only the translocation of activated ERK to the nucleus that is affected by Rho activation, other groups have shown that increases in ERK phosphorylation are also Rho-dependent (34–36). Of particular interest, a recent study suggests that Rho kinase signals through LIM kinase to produce stress fiber formation–dependent ERK phosphorylation, which in turn regulates the timing of cyclin D1 expression and cell cycle progression (34, 35).
Rho and Hypertension
Increased total peripheral vascular resistance, a hallmark of hyper-tension, results in part from enhanced agonist-stimulated blood vessel contraction (37–39). The contraction of resistance arteries mediated by α1 -adrenergic receptors is significantly greater in hypertension, whereas contraction stimulated by KCl (i.e., mediated by increased intracellular Ca 2+ ) is no different from the normotensive state (37, 38). These observations suggest that agonist- stimulated Ca 2+ -sensitization, a Rho-dependent process, may be enhanced in hypertension. This notion is further supported by evidence that serotonin-stimulated contraction of permeabilized coronary arteries at constant levels of Ca 2+ (i.e., reflecting Ca 2+ -sensitization) is increased in vessels from spontaneously hyper-tensive rats (SHR) (39). Another key finding implicating Rho in increased vascular resistance in vivo is that the Rho kinase inhibitor Y-27632 reduces blood pressure in three experimental forms of hypertension (27). More recently, Y-27632 was also shown to produce cerebral (basilar artery) dilation, which was significantly greater in SHR or rats made hypertensive by treatment with the nitric oxide synthase inhibitor L-NAME (N-nitro-L-arginine methyl ester) than in normotensive Wistar-Kyoto rats (WKY) (40). These studies highlight the importance of the Rho kinase pathway in blood pressure control and suggest a relationship between the extent of Rho expression/activation and blood pressure.
As indicated above, VSMC proliferation and migration are also Rho-dependent responses (1), which are likewise enhanced in hypertension. Both VSMC (41) and endothelial cells (42) from hypertensive rats have increased migratory capacity when compared to cells from normotensive rats. VSMC from SHR also exhibit a greater ability than control WKY cells to proliferate when cultured in serum or stimulated with AngII (43, 44). Hypertension is a clear risk factor for cardiovascular disorders such as restenosis, atherosclerosis, and renal failure, and VSMC proliferation and migration contribute to the pathophysiological progression of these vascular diseases. Indeed, clinical studies suggest that a number of antihypertensive therapies, although effective at lowering blood pressure, are not fully effective at reducing arteriosclerotic remodeling of resistance arteries, coronary events, and renal failure [see (45–47) ]. Thus, new molecular targets for pharmacological regulation of vascular proliferation and migration are being sought, and Rho kinase inhibitors and statins are clearly of interest for their therapeutic potential.
Both the expression of RhoA and the fraction of RhoA in the active state are increased in the vasculature of hypertensive rats; concomitantly, p27 Kip1 expression is reduced and rates of Rho-dependent DNA synthesis are increased (Figure 1⇓) (48). Additional work has established that there are increases in vascular Rho(A) activation in stroke-prone SHR, renal hypertensive rats, and deoxycorticosterone acetate (DOCA)-salt hypertensive rats (49, 50), as well as in models of diabetes (51, 52). These findings lend strong support to the hypothesis that deregulation of Rho activation and signaling contribute to hypertension and a variety of associated vascular pathophysiologies.

Elevated DNA synthesis and RhoA activation in spontaneously hypertensive rats. A. DNA synthesis in aortic smooth muscle cells from spontaneously hypertensive rats (SHR) and normotensive Wistar-Kyoto (WKY) rats. Cells were grown to confluence and serum-starved for 24 hours and then exposed to either vehicle, thrombin (Thr,12nM) or phenylephrine (PE, 10 μM) for 48 hours and labeled with [3H]thymidine for the last five hours prior to harvest for measurement of [3 H]thymidine incorporation into DNA. [See (45).] B. RhoA activation in aortic smooth muscle cells from SHR and WKY rats is measured as outlined in the text. Cells were stimulated with vehicle, thrombin (Thr,12nM) or phenylephrine (PE, 10μM) for 3 minutes. Cells were lysed and the lysates centrifuged at 14,000 x g for two minutes. The remaining supernatant was either used for Western blot analysis for total RhoA or added to a GST-fusion protein of the Rho binding domain of the Rho effector rhotekin precoupled to glutathione beads, followed by Western blot analysis for active RhoA.
The ability of GPCR agonists to effect vascular RhoA activation and/or signaling is also altered in experimental hypertension. For example, thrombin, which normally increases RhoA activation, is markedly more effective in this regard in aortic VSMC from SHR, relative to WKY rats (Figure 1⇑). Similarly, AngII is more effective in causing RhoA activation in preglomerular smooth muscle cells [SHR vs WKY rats (unpublished observations; E.K. Jackson, B.T. Andresen, T.M. Seasholtz, C. Zhu, and G.G. Romero)]. The ability of other GPCR agonists to increase RhoA activation or signaling is unmasked in VSMC from SHR: For example, whereas neither α1 - nor α2 -adrenergic stimulation affects RhoA activation, α2 agonists increase AngII-stimulated RhoA activation in SHR preglomerular smooth muscle cells (unpublished observations; E.K. Jackson, B.T. Andresen, T.M. Seasholtz, C. Zhu, and G.G. Romero), and α1 stimulation elicits Rho-dependent DNA synthesis in SHR aortic smooth muscle cells (Figure 1⇑). Thus, although there are modest but significant increases in RhoA expression in the SHR vasculature and VSMC, hypertension is primarily associated with the deregulation of the activation state of RhoA, along with a perturbation of agonist signaling (48–50). The observed changes in the ability of RhoA to be activated implicates alterations at the level of Rho GAPs, GEFs and/or GDIs. Neither the expression of Rho GDIs nor that of the leukemia-associated RhoGEF (LARG) appears to differ in VSMC from WKY and stroke-prone SHR (50) but possible alterations in the expression and function of other RhoGEFs or GAPs remain to be explored.
Rho and Nitric Oxide Synthase: Reciprocal Regulation and Endothelial Dysfunction
Discovery of the prominent role of nitric oxide (NO) as a vasodilator was a critical cornerstone in explaining the known relaxant effects of nitroprusside (i.e., a generator of NO) and in identifying the endothelium-dependent relaxing factor (EDRF) as NO (53, 54). Impaired endothelial function, resulting in decreased NO production, has been implicated in the etiology of both hypertension and atherosclerosis [see (55)]. Decreased endothelial nitric oxide synthase (eNOS) expression has been reported in aortas from adult SHR (56), and blood pressure is elevated in eNOS-deficient mice (57). The delivery of the eNOS-encoding gene into the tail vein of SHR, moreover, produces a significant reduction in blood pressure (58). There is thus compelling evidence that decreased eNOS is a mediator of deregulated blood pressure in hypertension. Interestingly, there is extensive crosstalk between the regulation of NO synthesis and Rho signaling pathways. A seminal finding in this regard is that the inhibition of Rho function by statins or exonuclease C3 upregulates NOS expression (18, 59, 60), and inhibition of Rho kinase results in increased NO production (61). These observations suggest that Rho serves to limit NO synthesis (59, 60). We have additionally found that VSMC express both eNOS and nNOS. Furthermore, Rho may decrease the expression of nNOS and subsequent NO production in VSMC (unpublished observation; T. Seasholtz).
In addition to effects of Rho on NOS, there is growing evidence that the NOS pathway can affect Rho function, in particular the membrane association of RhoA (62), Rho-mediated contractile responses, and RhoA activation (63–65). The ability of the NOS pathway to inhibit VSMC proliferation, migration, and contractility (66) may also result, in part, from the direct effect of NO diffusing from endothelial cells or of NO formed within VSMC to downregulate Rho signaling. Taken together, there is strong evidence for a signaling loop between Rho and the NOS pathway(s), in which increases in vascular Rho signaling might contribute to decreases in NOS, and decreased levels of NO further elevate Rho signaling (Figure 2⇓). Mechanistically, NOS activity may serve to protect against the enhanced contractile and proliferative changes associated with hypertension by decreasing the activity of RhoA.
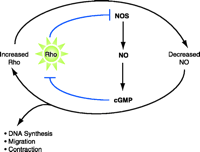
Hypertensive implications of Rho signaling. Rho decreases the mRNA stability of eNOS, which would thereby reduce NO production and subsequent cGMP-dependent signaling; a reduction of the inhibitory effect of cGMP on Rho activation would result in increased Rho signaling. In the schematic, arrows indicate processes of activation; blunt arrows represent inhibitory processes. Because the signaling scheme contains two blunt arrows, a positive feedback loop centers around Rho, resulting in contraction, migration, and DNA synthesis.
Inflammation, Vascular Injury, Atherosclerosis and Rho
Restenosis injury and atherosclerosis share a number of common pathological features. Whether through mechanical disruption of the endothelial layer or accumulation of lipid, the vascular wall undergoes local inflammation. Inflammatory mediators such as Tissue Necrosis Factor (TNF)-α and Interleukin (IL)1-β contribute to the upregulation of monocyte chemotactic peptide 1 (MCP-1) and adhesion molecules such as Vascular Cell Adhesion Molecule 1 (VCAM-1) and Intercellular Adhesion Molecule 1 (ICAM-1), which in turn lead to the recruitment and adhesion of monocytes to the vascular wall. Administration of antibodies against VCAM-1 following injury limits restenosis injury (67), consistent with the hypothesis that initiation of these early events leads to subsequent proliferation and migration of VSMC and vessel occlusion. A common therapeutic site of intervention for restenosis and atherosclerosis has recently emerged. Inhibitors of Rho kinase [e.g., Y-27632 and fasudil (i.e., HA-1077)] or expression of a dominant–negative form of the enzyme inhibits neointimal formation following vessel injury in a number of models of restenosis and reduces atherosclerosis (68–72). Furthermore, Rho kinase inhibition not only reduces lesion formation in low density lipoprotein (LDL) receptor knockout mice fed a high-fat diet (73), but also can limit cell proliferation and inflammatory processes (74). Statins, which can disrupt Rho localization and function (see above), have also been shown to inhibit VSMC proliferation following injury (75) and to reduce atherosclerotic lesion formation (76). Together, these studies suggest an important role of the Rho signaling pathway in vascular inflammation and responsiveness to injury in vivo.
Rho Activation by Mediators of Inflammation
A variety of humoral factors are formed during inflammation and injury. A number of these agents directly activate Rho, whereas others have been shown by more indirect means to signal through Rho pathways. Among these agents are certain GPCR agonists, including thrombin and sphingosine-1-phosphate (S1P), and cytokines such as TNF-α and IL1-β. Of additional interest is the possibility that LDL, which accumulates in atherosclerotic lesions and undergoes oxidative modification, likewise signals through Rho. It is likely that signals from a number of these stimuli converge to affect Rho-dependent cellular responses such as gene expression and cell proliferation.
GPCR Agonists
A subset of agonists for GPCRs are highly effective activators of RhoA in VSMC and other tissues. Prominent among these are thrombin, thromboxane A2 , and lysophospholipids such as lysophosphatidic acid (LPA) and S1P (1, 48, 77, 78). The receptors for these agonists couple to the G12/13 family of heterotrimeric G proteins. Seminal work by Sternweis and colleagues first elucidated the link between Gα13 and RhoA activation and demonstrated that Gα13 enhances nucleotide exchange on RhoA through its interaction with a specific GEF (i.e., p115 RhoGEF) (79). Subsequent studies have suggested that the G12/13 family of heterotrimeric G proteins are capable of interacting with other RhoGEFs (e.g., PDZRhoGEF, LARG and Dbl), and thereby activate Rho signaling pathways (80–82). A significant literature documents the acute (i.e., within minutes) activation of RhoA by GPCR agonists in a variety of tissues/cells; however, neither long-term effects of these agonists on RhoA expression and activation, nor the ability of GPCR agonists to regulate other Rho isoforms (e.g. RhoB and RhoC), have been established.
Cytokines
Cytokines such as TNF-α also elicit acute RhoA activation (83), and TNF-α stimulation of MAP kinase, endothelial permeability, and E-selectin expression have been shown to be inhibited by both Y-27632 and statins (84, 85). The mechanisms underlying the effects of TNF-α on RhoA remain to be determined, because there is to our knowledge no demonstrated link between molecules in the TNF-α signaling cascade and Rho GEFs or GAPs. One possibility is that TNF-α activates RhoA secondary to activation of sphingosine kinase, resulting in S1P formation and signaling (86, 87). Indeed, one report suggests that TNF- α–induced MCP-1 expression in response to oscillatory flow is mediated by S1P in human aortic endothelial cells (87). IL1-β also increases nucleotide exchange on RhoA and induces C3-sensitive actin stress fiber formation in HeLa cells (88); however, in smooth muscle cells, IL1-β treatment was found to decrease Rho activation (89).⇓

Rho signaling in vascular diseases. The upregulation of vascular RhoA, observed in hypertentsion, is hypothesized to enhance VSMC proliferation and inflammation in response to injury and atherogenesis. Inflammatory mediators including GPCR agonists, cytokines, and oxidatively modified LDL (oxLDL) signal through Rho to produce a variety of cellular responses involved in cell contraction, proliferation, and inflammation. These Rho-mediated responses contribute to the development of vascular diseases. Rho acting through Rho kinase has also been suggested to produce LIM kinase– and stress fiber–dependent activation of ERK, which in turn regulates the timing of cyclin D1 expression and cell cycle progression. It is likely that inflammatory mediators signal through Rho to activate NF-κB, increasing the expression of monocyte chemotactic peptide 1 (MCP-1), and adhesion molecules such as VCAM-1 and ICAM-1, leading to recruitment and adhesion of monocytes to the vascular wall. All of the signaling components mentioned above predispose to vascular disease.
LDL
LDL accumulates in the vascular wall during atheroma formation, and progression of inflammation leads to LDL oxidation Mounting evidence has shown that oxidatively modified LDL (oxLDL) promotes proliferation of VSMC (91) and endothelial cells (92), potentiates vasoconstrictor responses (93), and some-times downregulates eNOS expression (94). Although the mechanisms responsible for the cellular effects of oxLDL have not been established, components of oxLDL, such as LPA and lysophosphatidylcholine (93, 95), and generation of S1P (91, 96, 97) may be involved. Because LPA and S1P act primarily through Rho-mediated pathways, Rho is likely involved in cell growth responses to oxLDL. Indeed, there are several lines of evidence suggesting that Rho is activated by oxLDL: 1) oxLDL increases membrane-associated and decreases cytosolic RhoA in rabbit aortic ring homogenates (93); 2) oxLDL potentiation of AngII-induced vasoconstriction is reduced by Y-27632 and C3 (93); and 3) statins inhibit oxLDL-induced endothelial cell proliferation (92).
Is NF-κB a Mediator of Rho Signaling?
Nuclear factor κB (NF-κB) is a transcription factor that is activated in response to inflammatory stimuli. Under basal conditions, NF-κB is maintained in the cytosol by virtue of its association with κ-B inhibitory proteins. Upon stimulation, κ-B is phosphorylated by κ-B kinase (IKK) and dissociates from NF-κB; consequently, NF-κB enters the nucleus, where it can induce inflammatory genes such as those that encode MCP-1, VCAM-1, and ICAM-1. Phosphorylated κ-B is then polyubiquitinated and proteosomally degraded [ see (98, 99)].
Cytokines such as TNF-α and IL1-β are well-known activators of NF-κB, as are GPCR agonists that couple to G12/13 and Rho signaling pathways (100–102). Furthermore, there is recent evidence suggesting that both GPCR- and TNF-α–stimulated NF-κB activation depend upon Rho signaling. For example, fMet-Leu- Phe (fMLP; i.e., a GPCR agonist) stimulates expression of an NF-κB–sensitive reporter gene, and this can be prevented by over-expression of a dominant-negative form of RhoA (103); bradykin-mediated NF-κB activation is likewise blocked by inhibition of Rho by C3 (100, 104); blocking Rho function with exoenzyme C3 prevents TNF-α stimulated nuclear translocation of NκB (105). Despite evidence for Rho involvement, the me chanism by which Rho activates IKK and/or NF-κB has yet to be elaborated. Some studies have addressed the question of whether Rho signals through Rho kinase or another Rho effector to mediate NF-κB activation, but conclusions in the literature are conflicting (105, 106).
A link between NF-κB and VSMC proliferation is suggested by the observation that cytokine gene expression and VSMC proliferation are inhibited by overexpression of a proteosome-resistant form of κ-B (107). A separate study has demonstrated that the proteosome-resistant form of κ-B can reduce neointima formation following vascular injury (108). Because TNF-α and IL1-β increase VSMC proliferation and expression of MCP-1, VCAM-1, and ICAM-1, and because these responses are mediated in part through Rho and NF-κB signaling, these intermediates could be important therapeutic targets for vascular diseases.
Conclusions
Under conditions of vascular inflammation, diverse inflammatory mediators are released that activate RhoA. Multiple lines of evidence suggest that enhanced Rho activation is involved in hypertension and that Rho may act, in this context, to downregulate cyclin-dependent kinase inhibitors and NOS, as well as to activate ERK- and NF-κB–mediated gene expression. We hypothesize that increases in RhoA activity that occur in hypertension predispose to enhanced vascular proliferation, migration, and inflammation in response to injury and lipid deposition. Future studies utilizing transgenic and gene deletion technology will help determine whether increases in VSMC RhoA activity are necessary or sufficient to produce hypertension or mediate responses to vascular injury. In addition, human pharmacogenomic studies could identify polymorphisms in Rho signaling molecules or specific patients in whom Rho signaling is dysregulated. Numerous findings implicating Rho in vascular responses highlight the potential therapeutic importance of components of the Rho signaling pathway as targets for the treatment of vascular diseases.
Acknowledgments
TMS is supported by an American Heart Association National Center Research Program Scientist Development Grant. JHB is supported for the work on this project by NIH Grant GM 36927. The authors would like to thank Dr. James Liao (Harvard Medical School), and Dr. Yury Miller and Dr. Julie Radeff-Huang (UCSD) for their advice and comments regarding the manuscript.
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Joan Heller Brown, PhD, is Professor and Chair of the Department of Pharmacology at The University of California at San Diego School of Medicine.

Tammy M. Seasholtz, PhD, is Assistant Adjunct Professor in the Department of Pharmacology at the University of California at San Diego School of Medicine. Address correspondence to TMS. E-mail tseasholtz{at}ucsd.edu; fax 858-534-4337.

