Lighting a Backfire to Quench the Blaze: A Combined Drug Approach Targeting the Vanilloid Receptor TRPV1
The natural product capsaicin, the pungent constituent in hot peppers, has long provided a window into hyperalgesia, neurogenic inflammation, and thermoregulation (1, 2). Capsaicin functions to stimulate and, at longer times of exposure and at higher doses, to block the function of C-fiber sensory neurons, which normally act to detect noxious stimuli. Capsaicin has thus afforded a tool to delineate the roles of this sensory pathway in normal and pathological conditions and, through desensitization or “defunctionalization” of this pathway, capsaicin has provided a lead structure for evaluating the potential of this pathway as a therapeutic target. Unfortunately, a limiting toxicity of capsaicin has been the intense pain it induces upon initial application.
Recently, the depth of knowledge about the biology of capsaicin function has been complemented by the elucidation of its mechanism. Binding studies with the ultrapotent capsaicin analog resiniferatoxin demonstrated the existence of a specific receptor (3) and cloning of this receptor by the Julius group (4) revealed it to be a member of the transient receptor potential (TRP) channel family, representing the first member of the TRP vanilloid (TRPV) subfamily. The TRP channels are relatively nonselective cation channels, with the consequences of their activation largely reflecting the influx of calcium into the cells (5). Demonstration that TRPV1 fully accounts for the specific actions of capsaicin and resiniferatoxin has been provided by gene knockouts (6, 7).
These advances have driven intense and very promising efforts to find novel therapeutics targeted to TRPV1. TRPV1 antagonists have commanded the greatest attention. Following the identification of capsazepine, which showed that antagonism of TRPV1 function was possible (8), a wide diversity of structures have been identified, with promising candidates displaying nanomolar potencies, oral availability, and suitable pharmacokinetics (9–11). Whereas some antagonists predominantly antagonized TRPV1 stimulation in response to capsaicin, others more broadly blocked TRPV1 activation by temperature and low pH as well, presumably reflecting stabilization by the antagonist of the closed state of the channel (12, 13). This latter class of antagonists represents the focus for developmental efforts. The therapeutic advantages of antagonists, systemically administered, are their limited duration of action and ease of application. One disadvantage is that antagonists would only be expected to block stimulation of C-fiber neurons through TRPV1 but not through other mechanisms. An additional concern, still being assessed, is the possibility of adverse effects associated with systemic TRPV1 blockade. On the one hand, TRPV1-knockout mice show only quite modest abnormalities (14). On the other hand, antagonists initially induce hyperthermia, reflecting inhibition of the basal partial activation of TRPV1 occurring under physiological conditions (15). In addition, TRPV1 is being identified in a broad range of cells and tissues other than C-fiber sensory neurons, and its possible importance in these other sites remains, in general, to be defined (14, 16).
The complementary therapeutic strategy has been to use TRPV1 agonists at dosages and treatment schedules to achieve desensitization/defunctionalization of TRPV1 and C-fiber sensory neurons (17). Here, capsaicin has provided the paradigm, although it is now apparent that it is possible to appreciably dissociate the pungency of agonists (i.e., intense initial pain representing their limiting therapeutic toxicity) from their activity for desensitization/ defunctionalization. Dramatic examples of these compounds that possess reduced pungency but high desensitizing activity include resiniferatoxin (18) and olvanil (19). For example, resiniferatoxin is only a few-fold more potent than capsaicin in pungency but is 3–4 orders of magnitude more potent for desensitization (18). Desensitization/defunctionalization can be of very long duration, on the order of months, which represents an advantage for treatment of chronic conditions, such as bladder hyperreflexia (20) or cancer pain (21). To limit the extent of desensitization/defunctionalization, the agonist is typically applied in a localized setting. To deal with the initial pungency, local or general anesthetic may be used.
The recent report by Binshtok et al. (22) represents a conceptually simple but elegant refinement of this latter approach (Figure 1⇓). The authors evaluate both the cellular and whole animal effects of co-application of capsaicin and QX-314, an impermeant local anesthetic. The basis for the approach is that the local anesthetic will gain entry through the activated TRPV1 channel, thereby achieving blockade exclusively of the C-fiber neurons. The authors thus combine the specificity of action of a vanilloid agonist with the short-term blockade of a local anesthetic, avoiding the more complicated and potentially long-term desensitization/defunctionalization achievable at higher vanilloid agonist concentrations.
A core element underlying this approach was the report by Meyers and coworkers that HEK293T cells exogenously expressing TRPV1 rapidly took up the large cationic styryl pyridinium dye FM1-43 (cation MW = 452) upon TRPV1 activation by capsaicin or heat (23). Likewise, the cationic dyes MEQ (6-methoxy-N-ethylquinolinium, cation MW = 188) and MQAE [N-(ethoxycarbonylmethyl)-6-methoxyquinolinium, cation MW = 246)] can traverse the opened TRPV1 channel, with both rapid influx and efflux, under physiological conditions (24). It is important to note, however, that TRPV1 is not the only cation channel able to accommodate such large cations. Meyers and co-workers also described similar behavior for the purinergic type 2X (P2X2) channel upon stimulation by extracellular ATP and for the mechanotransduction channels of inner ear hair cells (23). Likewise, YO-PRO-1 (cation MW = 375) traverses channels formed by some members of the P2X channel family (25, 26). A further qualification is that, in mice injected with FM1-43, both large and small diameter neurons in the dorsal root ganglion (DRG) were labeled, indicating that labeling in these neurons was not limited to the small diameter TRPV1-containing neurons, and it did not require addition of an exogenous stimulus, suggesting that tonal activity of the channels was sufficient (23).
The second core element underlying the present report was the characterization, dating back to the 1970s, of the action of local anesthetics containing a quaternary amine group. Local anesthetics such as lidocaine block voltage-sensitive sodium channels. Substitution of the tertiary amine in lidocaine with a quaternary amine yields the derivative QX-314 (cation MW = 263). An important consequence of the presence of the quaternary amine group is that QX-314 maintains a positive charge and is, therefore, membrane impermeant. For the neuronal sodium channel, local anesthetics can only access the channel from the inner face of the membrane. QX-314 is therefore inactive unless it can somehow gain access into the cell (27). It should be noted that such selectivity is not true for all sodium channels. The cardiac sodium channels possess a second route for access of compounds such as QX-314, which can slowly enter the channel from the outside.
In the report by Binshtok et al. (22), the authors first demonstrate, using cultured rat DRG cells, that the combination of QX-314 and capsaicin induces a time dependent block of sodium currents, being virtually complete within 15 min. The effect was seen only in small but not large diameter neurons, consistent with the presence of TRPV1 only in the small diameter neurons. The authors then extended their analysis to two animal models of nociception. The first measured the threshold for paw withdrawal in response to pressure from von Frey hairs, a series of filaments of increasing stiffness. Capsaicin injection into the paw led to a sensitized response for 15–30 minutes. QX-314 by itself had no effect. The combination showed no early sensitization and a marked increase in the threshold between 1–3 hours, followed by recovery. The second model measured latency of response to noxious heat. Again, injection of capsaicin into the paw induced sensitization for 15–30 minutes; the combination of capsaicin and QX-314 induced no early sensitization and showed increased latency of response over the next four hours. Finally, the authors examined the effects in a more therapeutically analogous setting, using injection of the agents in the proximity of the sciatic nerve and then measuring the response of the animal to mechanical or thermal challenge as before. Again, capsaicin induced transient sensitization and the combination inhibited response to the noxious challenge with an onset at 15–30 minutes and a duration of an hour or so. Importantly, motor function was not affected. The authors conclude that the strategy they present for blocking pain should be advantageous for generating pain-restricted local anesthesia when preserving motor and autonomic responses and non-painful sensations is desirable or for treating nociceptor-driven chronic pain such as postherpetic neuralgia (i.e., pain associated with reactivation of a latent herpes virus family infection).
An important issue that remains to be resolved is the scope of the anti-nociceptive effect. On the one hand, C-fibers mediate hyperalgesia to thermal or, to a lesser degree, mechanical hyperalgesia, as has been shown by TRPV1 knockouts, by the action of vanilloid antagonists, or by destruction of C-fibers upon treatment of newborn mice with high doses of capsaicin. The expectation is thus for something less than pain-restricted local anesthesia. On the other hand, Meyers et al. (23) described the uptake of FM1-43 through a range of channels, not just through TRPV1, and, in the animal, into a range of neurons, including the large diameter neurons of the DRG that do not contain TRPV1. These results predict a broader spectrum of effect for QX-314. Perhaps consistent with this latter prediction, the results described by Binshtok et al. (22) for the increased threshold to stimulation by von Frey hairs contrasts with no difference between control and TRPV1-knock out mice under basal conditions in their response to mechanical stimulation, including by von Frey hairs, as reported by Caterina et al. (6) or to a distinct measure of mechanical nociception, as described by Davis et al. (7).
A second issue is the optimization of vanilloid and local anesthetic. Depending on the size of the TRPV1 channel, judicious choice of the size and nature of the local anesthetic (28) might provide enhanced selectivity, helping to address the concern raised by the results of Meyers et al. (23). Reciprocally, given the very different behavior of different vanilloid agonists on TRPV1 function in cells (29), it should be interesting to assess whether different vanilloids are differentially able to regulate permeability of large cations like QM-314 through the TRPV1 channel. Finally, it might be appropriate to explore whether, in pathological conditions, the vanilloid is actually essential for the action of QX-314. If the involvement of the C-fiber neurons is reflecting the physiological activation of TRPV1, then this activation itself might suffice to permit the entry of QX-314. If so, then inhibition might be even more specifically limited to only those C-fiber sensory neurons involved in the pathological response.
For millennia, vanilloids have been used in herbal medicine for the treatment of pain and other conditions (30) and their hotness is a matter of personal experience for almost all of us. Between the vigorous efforts in drug design and the rapid progress in the understanding of their mechanism of action, vanilloids present great promise for therapeutic application in the near future. The paper by Binshtok et al. (22) provides an exciting example of the rich opportunities for intervention in this system.
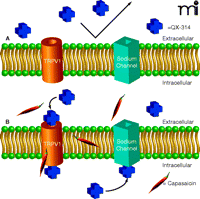
Combined treatment with capsaicin and positively charged sodium channel blocker to selectively inhibit C-fiber sensory neurons. A) The voltage-dependent sodium channel blocker QX-314 is unable to pass through the membrane on account of its positive charge. B) The vanilloid agonist capsaicin opens the TRPV1 channel, permitting the positively charged QX-314 to enter the cell through the relatively large pore of this channel. Upon gaining access to the interior of the cell, QX-314 inhibits the neuronal voltage-dependent sodium channel. Because TRPV1 channels are only present on the C-fiber sensory neurons, the action of the sodium channel blocker is thus limited to these neurons.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Peter M. Blumberg, PhD, is Chief of the Molecular Mechanisms of Tumor Promotion Section of the Laboratory of Cancer Biology and Genetics, Center for Cancer Research, at the National Cancer Institute. His research is focused on the biochemical pharmacology of TRPV1 and of protein kinase C. E-mail blumberp{at}dc37a.nci.nih.gov; fax 301-496-8709.

