S-Glutathionylation: Indicator of Cell Stress and Regulator of the Unfolded Protein Response
Abstract
The specific posttranslational modification of protein cysteine residues by the addition of the tripeptide glutathione is termed S-glutathionylation. This process is promoted by oxidative and nitrosative stress but also occurs in unstressed cells. Altered levels of S-glutathionylation in some proteins have been associated with numerous pathologies, many of which have been linked to redox stress in the endoplasmic reticulum (ER). Proper protein folding is dependent upon controlled redox conditions within the ER, and it seems that ER conditions can in turn affect rates of S-glutathionylation. This article seeks to bring together the ways through which these processes are interrelated and considers the implications of these interrelationships upon therapeutic approaches to disease.

Introduction
Cellular homeostasis is an intricate balance of survival and death signals, many of which are mediated by the posttranslational modification of proteins. Protein phosphorylation is probably the most widely appreciated example of such modification, whereby an interplay of kinases and phosphatases determines which hydroxyl-bearing side chains (i.e., Ser, Thr, and Tyr) of any given target protein are phosphorylated. New observations have made clear that the specific posttranslational modification of the sulfhydryl-bearing side chains (i.e., Cys) can likewise participate in the relay of cell signals. In particular, the S-glutathionylation of proteins, whereby a disulfide bond is established between a protein cysteinyl residue and glutathione (GSH), is, like protein phosphorylation, a reversible mechanism of protein regulation (Figure 1⇓). Significantly, S-glutathionylation can be promoted by physiological levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS), so that posttranslational modification of target proteins in this case is directly linked to the redox status of the cell. Indeed, the reaction can serve to protect proteins from oxidative or nitrosative damage and can also effect changes in protein structure that may be relevant to function or subcellular localization.
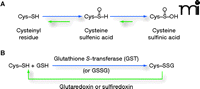
Reactivity of cysteinyl residues and the process of S-glutathionylation. A. The cysteinyl residues of proteins (Cys; Cys–SH to stress the thiol form) of cells under oxidative/nitrosative stress can be oxidized to various acidic forms, including cysteine sulfenic acid (Cys–SOH), which is fairly labile and can be readily reduced back to the thiol (Cys–SH) or further oxidized to the more stable cysteine sulfinic acid form (Cys–SOOH). It is becoming increasingly clear that such oxidation may be essential to normal deactivation-and-reactivation cycles of proteins and enzymes. B. The more familiar reaction of protein cysteinyl residues, namely, in the formation of disulfide bonds (e.g., as a manifestation of protein secondary structure), is related to the process of protein S-glutathionylation, which is increasingly recognized as essential to cellular behavior in health and in disease states. In the presence of physiological concentrations of glutathione (GSH), specific cysteinyl residues, by virtue of their position and reactivity within protein microenvironments, can undergo such modification through reactions with oxidized glutathione disulfide (GSSG) or by glutathione-utilizing enzymes such as glutathione S-transferases (GST). This posttranslational modification becomes reversible under catalysis that involves small redox proteins such as sulfiredoxin and glutaredoxin. (Oxidation is indicated in blue; reduction is indicated in green.)
The reversibility of protein S-glutathionylation, like the reversibility of protein phosphorylation, is critical to the regulatory role of the modification in cell signaling. In the case of S-glutathionylation, small cysteine-rich proteins such as glutaredoxin and sulfiredoxin function in catalytic reactions to provide a modification cycle that is suited to the control of regulatory pathways (1–3). Moreover, a number of phosphatases are subject to S-glutathionylation; in this way, the two modification pathways are interrelated.
Glutathione S-Transferases and Kinase Regulation
Glutathione S-transferases (GSTs) are a family of phase II detoxification enzymes that catalyze the conjugation of GSH to electrophiles through thioether linkages (4). GSTs also play a regulatory role in cellular signaling by associating with critical kinases involved in cellular responses to stress, apoptosis, and proliferation (5, 6). The soluble human GSTs can be divided into classes based on structural analysis (i.e., alpha, mu, pi, and theta classes); the first reported example of kinase regulation by a GST was in the inhibition of c-Jun aminoterminal kinase (JNK) by a pi class GST (pi GST) (5). JNK, a stress-activated kinase, has been implicated in pro-apoptotic signaling and may mediate the cytotoxicity of a variety of chemotherapeutic agents (7). The phosphorylation of JNK activates c-Jun, resulting in subsequent activation of downstream effectors. In unstressed cells, low JNK catalytic activity is maintained through its sequestration within a protein complex that includes pi GST, JNK, and c-Jun (Figure 2⇓). Under conditions of oxidative or nitrosative stress, however [during which all three of these proteins are S-glutathionylated (8–10)], the pi GST·JNK complex dissociates, so that JNK is free to act on downstream gene targets, whereas the pi GST undergoes oligomerization. Other GST isozymes seem capable of mediating similar regulatory interactions. For example, GSTA1-1 inhibits JNK signaling caused by either inflammatory cytokines or oxidative stress (11). The authors invoke a mechanism similar to that shown for the pi GST·JNK interaction (Figure 2⇓) and offer the premise that GST isozymes may overlap in substrate specificity.
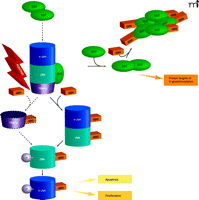
The interplay of protein phosphorylation and protein S-glutathionylation pathways in cell signaling. Under oxidative/nitrosative stress (indicated by ROS/ RNS), pi GST monomers associate with the target proteins c-Jun, JNK, and TRAF2, which are consequently S-glutathionylated (as indicated by –SSG). The immediate effect of S-glutathionylation could be TRAF2 activation, as well as the phosphorylative activation (indicated by –P) of c-Jun and JNK, culminating in the deployment of apoptotic or proliferative pathways. In addition, S-glutathionylation of pi GST results in its aggregation and concomitant inactivation. Dashed lines and arrows indicate hypothetical, rather than experimentally established, steps.
In addition, pi GST appears to regulate enzyme activities that otherwise manage cellular redox reactions. 1-Cys peroxiredoxin (1-cysPrx), which prevents oxidative damage to membrane constituents, can directly associate with pi GST (12, 13). During its catalytic cycle, a specific Cys residue within 1cysPrx undergoes oxidation to the sulfenic acid form, and the enzyme is thereby rendered inactive. Reactivation of oxidized 1cysPrx is effected by its heterodimerization with GSH-bound pi GST, leading first to the S-glutathionylation of 1cysPrx at its reactive Cys residue and subsequently to an intersubunit disulfide bond within the heterodimer. Finally, the GSH-mediated reduction of the disulfide bond regenerates the active, sulfhydryl-bearing form of 1cysPrx. Interestingly, the overexpression of 1cysPrx has been associated with cellular resistance to radiation as well as with suppression of JNK activation and apoptosis (14). Mutation of the catalytic Cys52 residue of 1cysPrx to Ser obliterates peroxidase activity and inhibits JNK activation; nevertheless, both wild-type and mutant proteins can be co-immunoprecipitated with pi GST and JNK. The authors concluded that 1cysPrx was able to suppress apoptosis through inhibition of JNK activation.
A further regulatory role for pi GST has been defined in the context of tumor necrosis factor-alpha (TNF-α) signaling. Wu and colleagues (15) showed that pi GST heterodimerizes with TNF receptor–associated factor 2 (TRAF2) and inhibits TRAF2-induced activation of both JNK and p38 (but not of NFκB). Class pi GST also interferes with the interaction between TRAF2 and apoptosis signal–regulating kinase 1 (ASK1), thereby inhibiting the autophosphorylation of ASK1 and apoptosis. Significantly, these effects, which are independent of the catalytic activity of pi GST, represent connectivity between S-glutathionylation and endoplasmic reticulum (ER) stress, because TRAF2/ASK1 activation mediates an ER stress–induced apoptotic pathway (see below).
Glutathione S-Transferases in Posttranslational Modification
In addition to its role in reactivating 1cysPrx (12, 13), pi GST may also catalyze the forward reaction of the protein S-glutathionylation cycle (Figure 1B⇑) (16). The rate and extent of protein S-glutathionylation is significantly reduced in pi GST-deficient animals, and in contrast to its inhibitory associations with 1cysPrx and TRAF2 (see above), the catalytic activity of pi GST is required for protein S-glutathionylation. HEK293 cells expressing catalytically inactive pi GST manifest diminished S-glutathionylation of cellular proteins in response to oxidative and nitrosative stress.
Interestingly, two Cys residues of pi GST itself are subject to S-glutathionylation (Cys47 and Cys101) (16), and the S-glutathionylation of pi GST reduces its enzyme activity against chemical substrates and promotes its multimerization. It is not known whether S-glutathionylation plays a role in the dissociation of pi GST from the kinases described above. However, Cys47 and Cys101 reside in distinct effector domains, both crucial for interaction with JNK, and Cys47 is critical for heterodimerization with 1cysPrx. In any event, a realistic model for pi GST in cell signaling will have to address both its enzymatic and inhibitory ligand roles in oxidative or nitrosative stress.
Targets of S-Glutathionylation
Although the S-glutathionylation of proteins was generally described in the 1990s, the identification of protein substrates was only made possible by the more recent advent of proteomic approaches. It is important to note that the actual number of cellular S-glutathionylated proteins is not large, relative to the proteome. Cluster analysis suggests that the modification may affect very specific cellular functions. In Table 1⇓, for example, six general categories of cell physiology appear to be affected by S-glutathionylation, each of which is basic to the biology of the cell: 1) the cytoskeleton; 2) metabolism and energy; 3) signaling proteins—particularly kinases and phosphatases; 4) calcium homeostasis; 5) protein folding and stability; and 6) redox homeostasis.
Cluster analysis of proteins susceptible to S-glutathionylation.
The most prevalent S-glutathionylated protein, and therefore most readily detected, is actin. When cells are stimulated with growth factors, S-glutathionylation of actin alters the ratio of soluble:polymerized protein. Consequently, there can be changes in the cellular architecture and membrane ruffling with concomitant changes in intracellular trafficking of many types of molecules. The S-glutathionylation of actin influences cellular adhesion and cell–cell interactions (17) as well as protein–protein interactions; for example, S-glutathionylated actin, relative to the unmodified protein, manifests a weaker affinity for tropomyosin (18).
Further elucidation of protein functions that are regulated through S-glutathionylation will likely carry clinical implications. For example, the constitutive activation of kinases has been intimately linked to cancer, with major impact upon drug development efforts. Similarly, the accumulation of S-glutathionylated proteins is emerging as a key factor in multiple diseases. Especially intriguing is the concurrence of increased protein S-glutathionylation rate and ER stress in the diseases highlighted in Table 2⇓. This overlap likely reflects signals emanating from the ER as it processes proteins that respond (e.g., by undergoing S-glutathionylation) to cellular redox conditions.
S-glutathionylated proteins identified in human diseases.
The Redox Environment of the Endoplasmic Reticulum
The ER is the first intracellular compartment for processing secretory and transmembrane proteins. Such processing may include a series of posttranslational modifications, notably glycosylation and disulfide bond formation, and depends on the distinct redox conditions provided within the ER. In contrast to the reducing conditions of the cytosol, where molar amounts of GSH may exceed those of GSSG by 100-fold, a highly oxidizing environment exists within the ER (GSH:GSSG ~ 3:1) to allow protein disulfide bond formation. This unique ER environment also provides the platform to sense oxidative and nitrosative stress. Protein folding within the ER is catalytically mediated, and chaperone proteins exist to prevent the aggregation of proteins as they undergo maturation within the ER (19). The folding process can be complex, involving multiple folding intermediates and the isomerization of disulfide bonds. (Proteins may also undergo reductive unfolding and subsequent degradation.) In yeast, molecular oxygen is the electron acceptor in the formation of protein disulfide bonds, which thus brings a risk for oxidative damage (20). In mammalian cells, it is well established that ER stress and the unfolded protein response (UPR; see below) are components of the hypoxic stress response in tumors (21). Clinical studies with the use of oxygen electrodes and markers of hypoxia have shown that O2 concentration heterogeneity occurs within individual tumors, ranging from zero to 100% (22). Translational attenuation occurs during hypoxia through the activation of pancreatic ER kinase (PERK), an ER resident kinase, and phosphorylation of eukaryotic initiation factor eIF2α. Cells derived from PERK-deficient mice are defective in eIF2α phosphorylation during hypoxia (23).
Multiple signaling pathways have evolved to ensure quality control in protein folding within the ER. Indeed, stress upon the ER results in the accumulation of malfolded proteins, leading to cellular deployment of the unfolded protein response (UPR). Specifically, the ER membrane contains three signal-transducing proteins that modulate the UPR: PERK, activating transcription factor (ATF), and IRE1 (Figure 3⇓). Regulation of these three proteins is contingent upon interactions with BiP [binding protein; also known as glucose related protein 78 (GRP78)]. The accumulation of malfolded proteins results in the dissociation of BiP and elicits the UPR, which culminates in three processes: 1) the inhibition of protein synthesis; 2) the increased expression of ER resident chaperones; and 3) the degradation of terminally malfolded proteins.
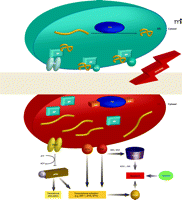
The Unfolded Protein Response (UPR) and pro-apoptotic pathways. The UPR is a complex signaling cascade that can be induced by stress and the accumulation of malfolded proteins in the ER. During homeostasis (upper schematic; blue), protein disulfide isomerase (PDI) promotes the proper folding of immature proteins (yellow strings). The ER-resident protein BiP associates with properly folded proteins and concomitantly inhibits three ER transmembrane proteins, namely, PERK, ATF6, and IRE1. Under conditions of oxidative and nitrosative stress (indicated by ROS/RNS and red background in lower ER schematic), PDI is modified (see text for discussion) and rendered inactive, and unfolded proteins (yellow strings) accumulate in the ER. BiP dissociates from improperly folded proteins and concomitantly surrenders negative regulatory interactions with PERK, ATF6, and IRE1. PERK thereby phosphorylates and inactivates (blunt arrow) eukaryotic translation initiation factor eIF2α, the phosphorylation of which is also associated with transcriptional activation of genes involved in the UPR. This transcriptional activation, which is also promoted by the activation of ATF6 and IRE1, can drive pro-apoptotic signaling, particularly through activation of CHOP (see text for details). IRE1 can also interact with TRAF2, which can function pro-apoptotically through association with ASK1 and JNK (see text; also see Figure 2). A third route to apoptosis is offered by caspase activity; intriguingly, regulation of caspase activity (particularly that of CASP3) may also be a function of GST.
The UPR can be viewed as a cascade of transcriptional and translational events that sense the capacity of the ER to manage the cellular demands of protein maturation. When the ER is unable to meet these demands, the UPR evokes apoptotic pathways (24). Three pathways (Figure 4⇓) for ER-induced apoptosis are known and can be triggered by ROS and RNS. The first involves activation of JNK and dissociation of the TRAF2–ASK1 complex. This pathway is redox-sensitive, and multiple proteins within this cascade are targets of S-glutathionylation. The second pathway involves transcriptional activation of the gene that encodes the C/EBP homologous protein [CHOP; also known as growth arrest– and DNA damage–inducible gene 153 (GADD153)]. The ER membrane proteins IRE1 and ATF6 are transcriptional activators of genes that encode ER-resident proteins and specifically activate the promoter of the CHOP-encoding gene. CHOP can be phosphorylated by the p38 MAP kinase family, leading to cell cycle arrest (25). Overexpression of CHOP leads to the proapoptotic translocation of BAX from the cytosol into mitochondria, whereas IRE1 has been shown to interact, via its cytoplasmic domain, with BAX as well as the related proapoptotic Bcl-2 member BAK and thereby to become activated (26). BAX and BAK have been shown to localize to mitochondria and the ER, where they presumably contribute to membrane permeability (27, 28). The third ER-induced apoptotic pathway involves caspase activation, including CASP3, a target of S-glutathionylation.
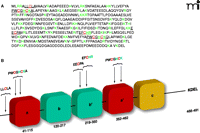
Regulation of PDI by its specific S-glutathionylation. A. PDI is a member of the thioredoxin superfamily of small thiol-rich proteins. The protein sequence of PDI includes seven cysteine residues (highlighted in red). Basic amino acids that flank cysteine residues (green) may act to lower the pKa of the thiol group, making the relevant Cys more likely to be susceptible to S-glutathionylation. B. A cartoon structure of the domains in PDI shows a single cysteine in the N-terminal domain, four in the catalytic domains, and two in the b’ domain.
Despite insights into the crucial roles of ER transmembrane proteins and BiP in the UPR, upstream events—especially the deregulation of chaperones that allows the accumulation of mal-folded proteins—are not well characterized. Human pathology and cancer drug discovery efforts have focused on protein disulfide isomerase (PDI), the most abundant ER chaperone, and its redox regulation as an upstream signaling event leading to the UPR (Figure 3⇑).
Protein Disulfide Isomerase
PDI is the most abundant chaperone in the ER. At 57 kDa, PDI is a large member of the thioredoxin superfamily and is organized into five domains (a, b, b’, a’, and c); it also contains a C-terminal KDEL sequence that targets it to the ER (Figure 4⇑) (29). PDI contains two active sites, in the a and a’ thioredoxin domains, each having two conserved cysteine residues that cycle between oxidized (disulfide) and reduced (dithiol) states (30). The crystal structure of yeast PDI suggests that the four thioredoxin domains (a, b, b’, a’) form a twisted U shape with the catalytic domains facing each other and an internal hydrophobic surface that interacts with malfolded proteins (31). Similar to pi GST, PDI has both enzymatic and protein binding functions. PDI facilitates the folding and correct S-S disulfide bond formation of its protein substrates. PDI is found in multimeric proteins such as prolyl-4-hydroxylase and microsomal triglyceride transfer protein (32, 33). PDI also interacts with the estrogen receptor to modulate its association with DNA (34, 35). PDI is also specifically and potently inhibited by estrogen, and PDI shares sequence similarity with the estrogen receptor. Numerous studies have demonstrated that PDI is key to ER homeostasis. PDI is regulated by the endoplasmic reticulum oxidase (ERO1), which restores reduced PDI to an oxidized state through disulfide exchange with ERO1. Interestingly, ERO1 activity is also regulated through modulation of noncatalytic cysteine residues (94). ERO1 activity is attenuated under oxidized conditions in the ER.
Posttranslational modifications that alter PDI function have recently been described. Nitrosylation of cysteine residues in the active sites of PDI occurs in the brains of patients manifesting sporadic Parkinson’s and Alzheimer’s disease (36), both of which involve ER stress and activation of the UPR. Nitrosylated PDI cannot function as a folding catalyst and thereby leads to the accumulation of malfolded proteins. In addition, PDI undergoes S-glutathionylation upon treatment with the anti-cancer agent PABA/NO (37), and we have shown that S-glutathionylation occurs at active-site Cys residues, leading to enzyme inactivation, activation of the UPR, and cancer cell death. Collectively, these studies provide evidence that redox-regulation of PDI is an upstream signaling event in the UPR.
Nitrosylation and S-glutathionylation of PDI are important posttranslational modifications, the deregulation of which can lead to tissue injury, with implications for cancer therapeutics. Proteomic analysis in a wide variety of cancer cell lines has shown alterations in the expression pattern of numerous PDI family members that correlate with differential drug response (38–41). The role of PDI in the cancer phenotype is not well characterized. A study with human breast ductal carcinoma tissue and histologically normal tissue concluded that a subset of approximately thirty proteins, which included PDI and six related proteins, were characteristic of epithelial neoplasia (42). A clinical correlation requires further confirmation; however, the UPR has been shown as a novel component of the hypoxic stress response in tumors and correlates with a poor drug response and more aggressive disease (42).
Drugs that Affect S-Glutathionylation
Early studies characterizing S-glutathionylated proteins used hydrogen peroxide as an inducer of oxidative stress. In subsequent studies, it became clear that a number of agents can produce oxygen or nitrogen radicals, and the definitions of ROS and NOS are quite broad. For example, ROS is associated with many radicals, including superoxide (O2.−), peroxyl (O2.), and hydroxyl (OH.) radicals and also encompass hydrogen peroxide and singlet oxygen (1O2). The definition of ROS is also broad as a result of the use of oxidized glutathione (GSSG) as a marker of an oxidizing cellular environment. Definitions of RNS include nitric oxide (NO.) and nitrogen dioxide (NO2−.) radicals as well as non-radicals such as nitrous acid, dinitrogen tetroxide (N2O4), and peroxinitrite (ONOO−). Endogenous ROS can be by-products of lipid peroxidation and the electron transport chain or caused by γ- and UV-irradiation. Elevated levels of nitric oxide (NO) provide the primary source of RNS. NO is an endogenous diffusible messenger shown to participate in survival and death pathways (43) and can alter protein function through direct modification or indirectly by generating products that ultimately lead to S-glutathionylation. Deregulation of endogenous NO production can lead to the release of RNS and ROS, each of which have been implicated in a number of human pathologies, including neuro-degenerative disorders, cystic fibrosis, aging, and cancer (44–46). This observation has led to investigations of the capacity of NO to induce cytotoxicity, with particular reference to antitumor activities (47), and resonates with the results and implications from work on GST (see above). It was the coalescence of NO and GST biology that led to the design and synthesis of the novel anticancer pro-drug PABA/NO that has been discussed above in the context of PDI inactivation (48).
The pi GST isozyme is expressed at elevated levels in a variety of human tumors and is linked with the development of resistance to a number of anticancer agents (46, 49). Catalytic activation of PABA/NO by pi GST releases NO that elicits anti-tumor activity both in vitro and in vivo (50–51). The pharmacology of PABA/NO predicts that NO will be quite rapidly released to result in extensive modification of target proteins. In fact, only two markedly nitrosated proteins were identified (3), whereas approximately twenty S-glutathionylated proteins were apparent (37). Although reactive cysteine residues are subject to both nitrosation and glutathionylation (e.g., PDI; see above), and although the identity of the direct glutathionyl donor may either be S-glutathione or a donor intermediate, the very fact that an NO-releasing drug causes such marked S-glutathionylation is in itself interesting. Nitrosated Cys residues may be susceptible to subsequent substitution by glutathione, which could help to explain the relatively small number of nitrosated proteins in PABA/ NO treated cells.
Adriamycin is an antitumor agent that is clinically used to treat solid and hematological tumors. The therapeutic value of the drug is tempered by dose-limiting toxicity that impairs macrophages and limits wound healing. Using cultured human macrophages, Asmis et al. showed that adriamycin causes S-glutathionylation of cellular proteins prior to caspase-independent death (52). These observations were validated in mouse models. Since anthracyclines such as adriamycin are known to cycle through redox reactions with quinone intermediates, it seems probable that free radical by-products of this drug may cause oxidative stress and thereby lead to S-glutathionylation. Whether these events have a bearing on the cytotoxicity of adriamycin is not firmly established.
Induction of oxidative stress may also occur by manipulation of the GSH:GSSG ratio. Recently, the novel cancer therapeutic NOV-002 was introduced to early clinical trial status in the US. The drug is a 1000:1 mix of GSSG and cisplatin, where the latter is not present in sufficient quantities to have a pharmacological impact. In common with a number of other sulfur-based therapeutics (53), NOV-002 has a stimulating effect upon myeloproliferation, a characteristic that underlies its therapeutic utility. In an in vivo setting, the drug can affect the GSH:GSSG ratio in blood and can stimulate S-glutathionylation, particularly of actin, although cell surface proteins are also S-glutathionylated in the presence of NOV-002 (54). Although NOV-002 does not induce apoptosis, the drug leads to ER stress. Neither the reduced nor the disulfide form of glutathione can passively cross the cell membrane; however, γ-glutamyl transpeptidase (γ-GGT) is an outer membrane–associated enzyme that catalyzes the cleavage of the GSH tripeptide from GSSG and glutathione conjugates and effects recycling of the constituent amino acids. A series of papers have proposed a mechanism whereby GSSG stimulates γ-GGT activity to result in the liberation of H2O2 into the intracellular environment [for review, see (55)]. Thus, extracellular exposure to excess GSSG could raise intracellular concentrations of ROS and thereby stimulate the S-glutathionylation of actin. Given the crosstalk between protein S-glutathionylation and phosphorylation pathways outlined earlier, GSSG (the active component of NOV-002) and γ-GGT could trigger the enhanced phosphorylation of regulatory kinase cascades that are involved in cell proliferation.
Conclusions
A variety of critical cellular processes are subject to regulation by protein S-glutathionylation. The list of modified proteins is not yet complete, nor is the structural and functional significance of the modification. Nevertheless, distinct crossover areas link phosphatase and kinase activity with S-glutathionylation, implying a control point between phosphorylation and thiol-mediated recognition of stress conditions. Certain drugs can promote S-glutathionylation and thereby affect a number of cellular functions, including proliferation and apoptosis. It is likely that the pathways affected are tissue-specific; in particular, aberrations of redox balance that have been identified in cancer may represent opportunities for therapeutic intervention. In cancer drug discovery, the UPR is an evolving target, and the indication that redox conditions in the ER may determine protein folding may have important implications for UPR-based clinical strategies. New approaches to diseases that involve alterations of cellular redox may benefit from the growing recognition of the role of S-glutathionylation in protein modification and the concomitant signaling events that elaborate the UPR.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Danyelle M. Townsend, PhD, is Assistant Professor in Pharmaceutical and Biomedical Sciences and Director of the Drug Metabolism and Pharmacokinetics Facility at the Hollings Cancer Center. She is interested in the molecular targets of ROS and RNS and cellular pathways that counteract stress. Her current work seeks to unite mechanistic and translational studies in the preclinical development of anticancer agents that target redox signaling. E-mail townsed{at}musc.edu; fax 843-792-9588.

