Splice Variants of GPCRs
- E-mail kminneman{at}pharm.emory.edu; fax (404) 727-0365
Abstract
Gprotein–coupled receptors (GPCRs) are encoded by a vast gene superfamily, reflecting the large number of ligands that must be specifically recognized at any given cell surface. The discovery that the variety of GPCRs is further expanded through the generation of splice variants was therefore somewhat surprising. Studies of the functional consequences of alternative splicing have focused on ligand binding, signaling, constitutive activity, and downregulation. However, GPCRs also appear to interact directly with many other intracellular proteins in addition to G proteins. Intriguingly, the domains involved in these interactions are the predominant sites of variation arising through splicing.

Each of the cultured
human cells shown above expresses the dopamine D3 receptor, a G protein–coupled
receptor, that has been manipulated so as to fluoresce red. Some of the cells additionally express a
truncated variant of the D3 receptor (green) that prevents trafficking of both variants to
the plasma membrane and that has been associated with schizophrenia. As discussed in the accompanying
article, other GPCRs are subject to less drastic forms of splicing that may play important roles in
their localization, trafficking, and signaling.
INTRODUCTION
G protein–coupled receptors (GPCRs) and ligand-gated channels are two important classes of receptors that help define cellular responsiveness to extracellular signals. GPCRs are structurally much simpler than the multimeric ligand-gated channels. It is believed that each GPCR contains a single binding site for its ligand, whereas ligand-gated channels manifest multiple ligand binding sites. Finally, splice variants of ligand-gated channels are common, whereas each GPCR is far more likely to be the single product of its gene. These qualities have tended to make GPCRs, one of the largest human protein families, attractive targets for drug development.
Recent studies, however, suggest that GPCRs may be more complex than previously thought. Evidence is now very strong that GPCRs: i) sometimes arise through alternative splicing; ii) may associate to form homo- or heterodimers within the cell membrane; and iii) can interact directly with other proteins in macromolecular complexes. These new perspectives have had a major impact on our understanding of GPCR pharmacology. In this article, I discuss splice variants of GPCRs and their possible functional significance, a topic recently reviewed by Kilpatrick et al. (1).
GPCR GENESTRUCTURE AND SPLICE VARIATION
Molecular cloning of GPCRs in the 1980s led to the realization that their genes have unusual structures. Although less than five percent of human genes lack introns in their protein-coding regions, most mammalian GPCRs (>90%) are intron-less (2). This finding led to the widespread use of genomic libraries for the cloning of additional of GPCRs, so that the possibility of alternative gene splicing was generally ignored. However, splice variants of the rat D2 dopamine receptor were first reported in 1989 (3), and other GPCR genes have since been found to contain introns in their open reading frames (ORFs).
It is now clear that the genes that encode GPCRs are structurally heterogeneous. As Table 1⇓ shows, however, an increasingly complex genomic structure does not correlate with the number of known splice variants. Although most GPCR genes have no introns in their ORFs, others (e.g., rhodopsin) have multiple introns and yet still appear to evade alternative splicing. Some GPCR genes, like that for the D2 dopamine receptor, have many introns but yield few splice variants, and others, such as that for α1A-adrenergic receptors, have one intron in their primary ORF but yield multiple splice variants. At the extreme, the GABAB-R1 (γ-aminobutyric acid type B receptor 1) gene has twenty introns in its ORF, with only two splice variants reported to date. Further comparison of GPCR genes must remain limited, because there are undoubtedly GPCR splice variants not yet identified. However, many GPCRs have been examined extensively and identification of large numbers of additional splice variants is unlikely.
Splice Variation and Complexity of GPCR-EncodingGenes
Why do most mammalian GPCR genes lack introns? In C. elegans, virtually all GPCR genes, which make up almost five percent of the organism's total gene content, contain introns in their ORFs (2). Similarly, serotonin 5HT1 receptor genes have introns in Drosophila (4), whereas the human 5HT1 receptor genes do not (5). It may be evolutionarily advantageous for the GPCRs in higher organisms to be encoded by intron-less genes (2). If so, human GPCR genes that do contain introns must be under pressure to retain them, possibly implying important functional differences between GPCR splice variants. It is interesting that all five human muscarinic acetylcholine receptor genes are intron-less, although the homologous C. elegans gene contains introns and encodes three splice variations within the third intracellular loop (6). Whether the increased number of GPCR genes in higher organisms is somehow related to decreased gene complexity is not yet clear.
Splice variants are usually identified by use of the reverse transcriptase–polymerase chain reaction (RT-PCR), or by parallel sequencing of multiple cDNA clones. Tissue distribution of splice variants is often supported by analysis of RNA (usually, in situ hybridization); however, their biological significance can be difficult to determine. Selective antibodies are not usually available, and the heavy reliance on PCR techniques and sequencing can complicate the interpretation of functional relevance. Premature stop codons that would produce proteins lacking one or more transmembrane domains have been reported for several GPCRs (1); however, it is not yet clear whether these truncated proteins are expressed, and they are not considered below. This review focuses only on splice variants of human origin that have been reported by at least two laboratories, or which have been confirmed by at least two methods. These criteria substantially reduce the number of splice variants considered (1) but increase the likelihood of functional relevance. The resulting survey shows that human GPCR splice variants cluster into three main groups (Figure 1⇓): those with variation in the intracellular C-terminal tail; those varying in the third intracellular (i3) loop; and those that vary in the extracellular N terminus. For reasons of space, the few splice variants that have been reported in extracellular and other intracellular loops (1) are not discussed here.
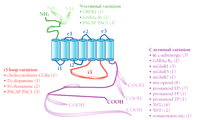
Schematic simplification of the GPCR structure and summary of three classes of human GPCR splice variants. The seven transmembrane domains are shown as blue cylinders; the remaining canonical polypeptide sequence is shown as colored string: extracellular loops (e1, e2, and e3), and two intracellular loops (i1 and i2) are shown in blue; the N-terminal sequence is green; intracellular loop i3 is red; and the C-terminal sequence is purple. Alternative splice variants, including those that represent alternative N and C termini, are indicated by the pod-like expansions superimposed upon the string. Numbers in parentheses next to receptor names indicate how many splice variants exist in addition to the first receptor cDNA sequence cloned. Human sequences verified by two independent labs or approaches are included. References for each of the splice variants are available in the online version of this article.
Splice Variants of the C-Terminal Tail
The largest group of splice variants pertains to the C terminus, and comprises thirty-four variants of twelve different receptors (Figure 1⇑). C-terminal splice variants of GPCRs share sequence identity with regard to their seven transmembrane domains, but differ in the length and sequence of the intracellular tail. Five of the receptors in this group have shown only a single splice variant, two show two splice variants, and five are more extensively spliced. These five receptors (α1A-adrenergic, metabotropic glutamate mGluR1, mu-opioid, prostanoid EP3, serotonin 5HT4) account for more than two-thirds (25/34) of the total C-terminal variants and more than half (25/46) of all verified human splice variants. Receptors that manifest splice variation in the C terminus can be further classified according to where and how their sequences diverge: Some show complete divergence shortly after the seventh transmembrane domain (i.e., mGluR1 and the prostanoid EP3, FP, and TP receptors); others diverge close to the C terminus (i.e., α1A-adrenergic, mu-opioid, 5HT4, mGluR7, GABAB-R2c); a final group results from a deletion (i.e., GABAB-R2b) or insertion (i.e., 5HT7, mGluR5) of a small sequence within the C-terminal tail (Figure 1⇑).
Splice Variants of the Third Intracellular Loop
Seven variants pertain to alteration of the third intracellular (i3) loop, representing four receptors (Figure 1⇑), and arise through variable insertion of coding exons. The dopamine D2 receptor, the first GPCR for which a splice variant was reported, exists in “short” and “long” forms that differ by twenty-nine residues in the i3 loop. The PACAP (pituitary adenylate cyclase activating polypeptide) receptor may be the most interesting example in this class, where variable insertion of two alternative 28-residue segments in the i3 loop results in four separate human isoforms. Similar splice variants of histamine H3 and cholecystokinin B receptors have also been reported.
Splice Variants of the N Terminus
Five variants of three receptors are confirmed that represent variation in the extracellular N terminus (Figure 1⇑). These are formed by exon skipping, resulting in either a truncated terminus, or a short deletion within the domain. Human GABAB-R1 receptors belongs to this group, with GABAB-R1b receptors having a truncated N terminus and GABAB-R1c receptors having a shorter deletion in this region. Similar isoforms have been reported for the CRFR2 (corticotropin-releasing factor receptor type 2) receptor and the PACAP receptor.
FUNCTIONAL DIFFERENCES BETWEEN SPLICE VARIANTS
Because there are often many closely related GPCRs for a particular ligand, the role of additional splice variants is unclear. For example, there are three α1-adrenergic receptor subtypes among the nine adrenergic receptors. These closely related subtypes respond to the same transmitter and activate the same signals (7), and so the existence of three additional splice variants of the α1A subtype is puzzling (8, 9). However, the structural regions where the splice variations occur have led to some ideas about their function. The i3 loop and C-terminal tail are often important in G protein coupling, whereas C-terminal phosphorylation plays an important role in desensitization. Thus, G protein coupling and desensitization of GPCRs alternatively spliced in these regions have been carefully compared. The N terminus is often involved in amino acid and polypeptide binding, and N-terminal splice variants have been proposed to influence binding specificity. In addition, recent findings about interactions of GPCRs with other proteins may provide new insights into the functional roles of splice variants.
Coupling Efficiency
G protein activation is the major function of GPCRs, and the efficiency and specificity of coupling of splice variants is worthy of particular attention. Coupling differences have been reported for several known splice variants, particularly for those differing in intracellular domains. When rat PACAP receptor isoforms with alternatively spliced i3 loops were first identified, they were shown to differentially couple to adenylate cyclase and phospholipase C (10). Similarly, these signaling pathways are also differentially affected by C-terminal splice variants of bovine prostanoid EP3 receptors (11), with two isoforms inhibiting and two activating adenylate cyclase. These data are consistent with previous ideas about the role of the i3 loop and C terminus in G protein coupling, and suggest that alternative splicing of GPCRs might be a major mechanism for controlling signaling specificity.
Nevertheless, differences in coupling between splice variants seem to be neither robust nor consistent. For example, human PACAP receptor splice variants differ from their rat orthologs in that distinct human isoforms show similar activation of adenylate cyclase (12). Similarly, all four human EP3 receptor splice variants inhibit adenylate cyclase (13), whereas a mixture of inhibition and stimulation occurs upon activation of the bovine isoforms. Rat SSTR2 (somatostatin receptor type 2) C-terminal splice variants do not show the differences in coupling observed with mouse orthologs (14), and the proposed differential coupling of i3 loop variants of the D2 dopamine receptor to distinct Gi proteins is also controversial (15).
These discrepancies may be due to the difficulties involved in comparing coupling efficiencies of closely related receptors. Heterologous expression is usually required, and the lack of suitable radioligands or antibodies can make it difficult to determine whether similar expression levels are achieved, and whether receptor density is within a physiological range. In fact, in studies where receptor density can be controlled and quantified, splice variants often show few differences in coupling efficiency. For example, human α1A-adrenergic receptor isoforms show comparable activation of phospholipase C (16), and human 5HT4 receptor isoforms cause similar activation of adenylate cyclase (17). Thus, although coupling efficiency may vary for alternatively spliced GPCRs, these differences are often subtle and their biological significance remains to be determined.
Desensitization
Phosphorylation of the C-terminal tail is important in the desensitization of many GPCRs; intriguingly, one of the most obvious differences among C-terminal splice variants is in the number of potential phosphorylation sites. For example, the C-terminal tail of the human prostanoid TPβ receptor contains an additional eleven serines, an additional four threonines, and one extra tyrosine compared to the shorter TPα isoform (18), and the mGluR1a contains fifty-four more Ser/Thr residues than the much shorter mGluR1b (19). For this reason, desensitization patterns of several C-terminal splice variants have been carefully compared.
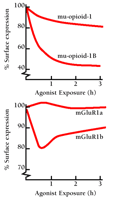
Despite these dramatic sequence alterations, differences in desensitization of C-terminal splice variants have been found to be relatively modest. The clearest differences appear to be associated with receptor internalization. As shown in Figure 2⇓, rat mu-opioid (20) and human mGluR1 (21) receptor C-terminal splice variants both exhibit differences in agonist-induced internalization. In both cases, chronic agonist exposure causes a greater loss in cell surface localization for the shorter isoform (mu-opioid-1B, mGluR1b) than for the longer isoform. These observations must be interpreted cautiously, however, because loss in receptor density at the cell surface is a function of both internalization and recovery to the membrane, two processes that can vary independently (20). Also, some C-terminal splice variants show no apparent differences in desensitization. Human prostanoid TPα and TPβ C-terminal isoforms have large differences in the number of potential phosphorylation sites, but a thorough study using isoform-specific antibodies found similar dose- and time-dependence for agonist-induced phosphorylation, and identical rates of desensitization (18). Other studies of the same receptors have, however, reported differences in splice variant internalization (22). Thus, C-terminal splice variants of GPCRs sometimes, although not always, show differences in desensitization.
Agonist-Independent Constitutive Activity
Some GPCRs spontaneously assume an active conformation without agonist binding, and differences in such constitutive activity have been reported for several splice variants. These differences have been found mainly with C-terminal splice variants, including mGluR1 (23), prostanoid EP3 (24), and 5HT4 receptors (25). An increase in the density of Gs-linked 5HT4 receptors from 100 to 500 fmol/mg protein caused an almost fourfold increase in basal cAMP production for the 5-HT4a splice variant, and other C-terminal splice variants were associated with altered rates of cAMP production (25). Differences in constitutive activity were also reported for C-terminal variants of the Gi-coupled prostanoid EP3 receptor, at least at elevated expression levels (24). Similar differences have been reported for Gq-coupled mGluR1 receptors, although receptor density could not be quantified (23). However, constitutive activity is difficult to quantify, and is generally inferred from differences in second messenger levels caused by changes in receptor density obtained by heterologous overexpression. The functional significance of such differences thus awaits further study.
Ligand Binding
Because intracellular domains do not generally contribute to formation of the ligand-binding pocket, it is not surprising that splice variation of intracellular sequences elicits almost no effect on ligand binding. Pharmacological differences are more likely to be associated with N-terminal splice variants. In fact, an N-terminal splice variant of the PACAP receptor, with a 21-residue deletion, causes a substantial change in agonist affinity (26). Conversely, no significant differences in ligand binding profiles have been reported for N-terminal splice variants of the CRFR2 and GABAB-R1 receptors. However, it is difficult to draw broad conclusions because the number of N-terminal splice variants is limited, and there are few drugs available for characterizing these receptors.
LOCALIZATION
Sequence-specific probes have made it possible to map, by in situ hybridization, the distribution of differentially spliced GPCR mRNA, particularly in brain. Not surprisingly, GPCR splice variants often show differential distribution among many tissues and brain regions (1), consistent with cell-specific control of transcription and splicing. A recent report suggests that different D2 dopamine receptor isoforms, formed by alternate splicing of the i3 loop, effect distinct functions based on their preferential pre- versus postsynaptic localization. Specifically, the targeted ablation of the additional exon that establishes the D2 long isoform resulted in mice expressing only the short form of the D2 receptor but evidencing normal total D2 receptor density and pharmacology. Significantly, however, D2-mediated postsynaptic motor responses were essentially abolished in these mice, whereas D2 presynaptic regulation of dopamine release appeared to be normal (27). Thus, the D2 long isoform functions primarily postsynaptically, and the D2 short isoform appears to be primarily presynaptic. Little is known about the mechanisms underlying such differential distribution, which may reveal quite a bit about the functional roles of these splice variants.
PROTEIN-PROTEIN INTERACTIONS INVOLVING INTRACELLULAR DOMAINS OF GPCRs
It has become clear in recent years that GPCRs exhibit specific interactions with many proteins in addition to G proteins, such as anchoring molecules, molecules involved in intracellular signaling, and “adaptor” molecules that may link receptors to other molecules or signals. Such protein–protein interactions depend mainly upon intracellular domains, particularly on the C-terminal tail (28). Because these are the domains most commonly altered in GPCR splice variants, it is likely that splicing will influence such interactions.
Although GPCRs have been known for many years to bind arrestins and kinases involved in desensitization, the diversity of other proteins that bind to GPCR intracellular domains has only recently become apparent. In this regard, one important protein is Homer, an immediate-early gene product found to associate with the C-terminal tails of mGluRs 1 and 5 (29). Homer represents a family of proteins, the longer forms of which have coiled-coil domains that mediate the formation of multimeric complexes. Homer proteins bind to a C-terminal proline-rich sequence of mGluR1 family members that is also present in both the ryanodine receptor and the inositol 1,4,5-trisphosphate receptor. It has been suggested that Homer proteins create a regulated physical link between mGluR receptors and inositol 1,4,5-trisphosphate receptors, and that this link is disrupted by production of the immediate-early-gene product Homer-1a (30), which lacks the coiled-coil domain.
Significantly, the Homer-binding motif is present in both splice variants of the mGluR5 receptor and in the mGluR1a receptor, but is not present in the three mGluR1b, mGluR1c, or mGluR1d receptor splice variants (31). In these latter three isoforms, the long C-terminal tail (318 residues) of mGluR1a is replaced by a much shorter C-terminal sequence (11-26 residues), eliminating the Homer-binding domain. If the current model is correct, removal of this binding domain will have an important impact on the properties of mGluR1 splice variants, probably leading to alterations in localization, coupling, and regulation.
Another novel protein found to interact with a GPCR intracellular domain is the sodium/hydrogen exchange regulatory factor (NHERF). This PDZ domain–containing protein associates specifically with the last four amino acids of the C terminus of the β2-adrenergic receptor in an agonist-dependent manner (32). PDZ domains are important components of molecular scaffolds that hold large multiprotein signaling complexes together, and are thought to correctly position signaling molecules for interaction. Although there are noβ 2-adrenergic receptor splice variants, there may be similar interactions of PDZ domains with other GPCRs. This is particularly interesting because C-terminal splice variants often differ by only a few amino acids at the end of the tail, precisely where PDZ domains bind.
Protein partners that interact with the i3 loop of GPCRs have also been identified (28). An example is spinophilin, which binds to the i3 loop of dopamine D2 receptors (33), where alternative splicing can occur. Protein–protein interactions may also be important in the function of N-terminal splice variants. The N-terminal domain of the GABAB-R1 receptor contains two complement protein modules (SCR or “sushi” domains) that are involved in specific protein–protein recognition events. Either one or both of these modules are removed in the two known human GABAB-R1 splice variants (35, 36). The functional significance of this splice variation has yet to be determined.
These observations suggest that important functional differences between splice variants may be explained by the presence or absence of protein-binding motifs and resulting differences in protein–protein interactions. Differential interactions of such proteins with GPCR splice variants could have a marked impact on their localization and/or functional properties, as illustrated schematically in Figure 3⇓. In fact, C-terminal binding partners for SSTR2 (SSTRIP/Shank1) (37), mGluR7 (calmodulin) (38), and α1A-adrenergic receptors (neuronal nitric oxide synthase) (39) have been reported. All of these receptors exist as C-terminal splice variants, although we do not yet know whether the partner proteins will bind differentially to these splice variants and the functional significance of such interactions remains unclear. However, the identification of such binding partners strengthens the hypothesis that alternate splicing of GPCRs may remove or add particular protein-binding motifs, resulting in receptors with differential localization and function.
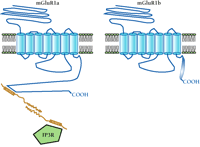
Potential role of protein–protein interactions in the differentiation of splice variant function. C-terminal splice variation may result in distinct protein-binding motifs, leading to specific interactions with intracellular proteins. This mode of functional regulation, mediated by splicing, is shown in the schematic for two variants of mGluR1. Specifically, the immediate-early gene product Homer is schematized to interact with mGluR1a but not the shorter C-terminal splice variant mGluR1b. Homer proteins bind to a proline-rich consensus motif in the C-terminal tail of mGluR1a that is spliced out of mGluR1b. The homodimerization of Homer through its coiled-coil domains is proposed to physically link the mGluR1a receptor to the inositol 1,4,5-trisphosphate receptor (IP3R), which also contains a Homer binding motif. Because the mGluR1b (shown), mGluR1c, and mGluR1d C-terminal splice variants do not contain a Homer binding motif, they would not be expected to associate with Homer proteins. Whether these C-terminal splice variants specifically interact with other proteins is not yet known. Other protein-binding sequences that may be introduced into or excluded from GPCRs are discussed in the text.
DIMERIZATION
Another surprising discovery was the recent observation that heterodimerization is necessary for correct assembly and function of some GPCRs. The major breakthrough came with the observation that the dimerization of distinct polypeptides, each containing seven transmembrane domains, was required to form a GABABR with native pharmacological and signaling properties (35). Heterodimerization is apparently required for export of one of the GABAB-R subunits to the cell surface and specifically functions to mask a C-terminal signal for retention in the endoplasmic reticulum (40). Interestingly, splice variation has been reported for GABAB-Rs both in the intracellular tail, thought to be responsible for dimerization (GABAB-R2), and in the extracellular N-terminus responsible for GABA binding (GABAB-R1). However, the subdomains that are alternatively spliced are not those currently believed to be involved in either dimerization or binding, and no effect of alternative splicing has yet been reported for either of these parameters. Analogous results have been reported for several other GPCRs, including opioid receptors, suggesting that GPCR heterodimerization may be relatively common (41). If other GPCRs with C-terminal splice variants also prove to undergo C-tail–dependent dimerization, important functional implications for the existence of splice variants will have to be considered.
CONCLUSIONS
It is now clear that a relatively small subset of GPCRs undergo alternative splicing to generate isoforms differing in particular intracellular and extracellular domains. Most of the verified human splice variants represent variation of GPCR C termini, although splicing can also affect other regions. We still know little about the functional roles of GPCR splice variants, but differences in ligand binding, signaling efficiency, constitutive activity, and desensitization have been reported. This area has recently become more interesting not only by the discovery of a large number of intracellular proteins that specifically associate with GPCRs, but also by the recognition that some GPCRs require heterodimerization for correct assembly and function. Because these protein–protein interactions often involve C-terminal domains, splice variants may arise as a mechanism to regulate such associations. The addition or deletion of discrete protein binding motifs by alternative splicing may thus confer important functional characteristics on particular splice variants, and the elucidation of such characteristics may provide new insights into the physiological function of GPCR splice variation.
- © American Society for Pharmacology and Experimental Theraputics 2001
REFERENCES

Kenneth P. Minneman, Ph.D., is Charles Howard Candler Professor of Pharmacology at Emory University. His research interests include the pharmacology, structure, molecular biology, and mechanisms of signal transduction of adrenergic receptor subtypes.

