Pharmacology of Capacitative Calcium Entry
- James W. Putney Jr., Ph.D.
- Laboratory of Signal Transduction National Institute of Environmental Health Sciences/NIH, PO Box 12233 Research Triangle Park, NC 27709
- E-mail putney{at}niehs.nih.gov; fax 919-541-7879.
Abstract
The versatility of Ca2+ as a messenger in multiple signaling events requires that the concentration of calcium ions within the cytoplasm be highly regulated. In particular, the release of calcium from intracellular stores must often be linked to calcium influx across the cell membrane. Capacitative calcium entry, whereby the depletion of intracellular Ca2+ stores induces the influx of extracellular calcium, is a crucial element of concerted calcium signaling. Investigations into the phenomenon are contributing to a new appreciation for the organized cytoplasmic framework that supports calcium signaling.
The endoplasmic reticulum (stained red above) is an important source of the calcium ions that pour into the cytosol in response
to extracellular stimuli. The mechanism by which release of calcium ions from intracellular stores is coordinated with ion
influx across the plasma membrane is referred to as capacitative calcium entry. Research into this mechanism will likely result
in new pharmacological approaches to cell signaling.
INTRODUCTION
Many key biological processes are regulated by cytoplasmic calcium signals. Such signals can arise either from the release of stored calcium or the influx of calcium across the plasma membrane, but more characteristically, from both routes. A common mechanism by which such cytoplasmic calcium signals are generated involves receptors that are coupled to the activation of phospholipase C (1). Phospholipase C generates inositol 1,4,5-trisphosphate (IP3), which in turn mediates the discharge of Ca2+ from intracellular stores. The fall in Ca2+ concentration within the lumen of the Ca2+-storing organelles (most commonly, components of the endoplasmic reticulum) activates plasma membrane Ca2+ channels. The retrograde process by which plasma membrane calcium channels are signaled by the endoplasmic reticulum has been called “capacitative calcium entry,” or “store-operated calcium entry” (2, 3).
Recent research has focused both on identifying components of the plasma membrane channels that underlie capacitative calcium entry, and on elaborating the mechanisms by which they respond to the depletion of calcium from within the endoplasmic reticulum. Although the components of the capacitative calcium entry channels have not yet been isolated, leading candidates are products related to the Drosophila trp (transient receptor potential) gene family (4).
Hypotheses about the signaling mechanism of capacitative calcium entry include the release of a putative calcium influx factor (CIF) from endoplasmic reticulum (5, 6), as well as the direct interaction of IP3 receptors in the endoplasmic reticulum with plasma membrane channels in a process called “conformational coupling” (7, 8). A number of recent publications have reviewed the evidence, controversies, and contradictions that characterize the mechanisms hypothesized for capacitative calcium entry (3, 4, 8-11). The current review will focus primarily upon the physiological roles of capacitative calcium entry, and will discuss current knowledge about the pharmacology of this important signaling mechanism.
THE PHYSIOLOGY OF CAPACITATIVE CALCIUM ENTRY
Capacitative calcium entry is one of the most prevalent mechanisms of cellular Ca2+ signaling and, in the cell types in which it occurs, plays one or more of three general roles (3). First, capacitative calcium entry replenishes cellular Ca2+ stores at the rapid rates required, for example, following transient receptor activation by neurotransmitters. Capacitative calcium entry is uniquely fitted to re-establishing cell readiness in this regard, because it assures that even after the initial stimulus is removed—and removal must often occur rapidly in neurotransmission—calcium influx across the plasma membrane can proceed until intracellular stores of Ca2+ are completely replenished. Second, capacitative calcium entry can provide prolonged, sustained elevation of intracellular Ca2+ concentration. Such sustained elevation, which may be desirable when tonic responses are needed, for example, in maintenance of smooth muscle tone, cannot be readily achieved without calcium influx, because intracellular stores can provide elevated concentrations for only a few minutes at most. Third, wherever repetitive baseline spike oscillations operate to sustain intracellular Ca2+ signals (12), capacitative calcium entry provides a Ca2+ cushion to maintain the constant amplitude of each spike in intracellular Ca2+ concentration. When internal Ca2+ concentration undergoes periodic spiking, it is important to coordinate the influx of Ca2+ across the plasma membrane with intracellular release to provide just the amount of Ca2+ needed to refill the stores. If Ca2 influx is excessive, baseline intracellular Ca2+ levels will gradually increase; if influx is insufficient, spike height and frequency will gradually decrease. The necessary balance is achieved because, during the interspike interval, capacitative calcium entry and its deactivation are coordinated to the charge of intracellular Ca2+ stores. In cells that do not manifest capacitative calcium entry, the replenishment of calcium stores is most likely accomplished by means of the plasma membrane sodium-calcium exchanger (13).
Capacitative calcium entry appears to be the major means of regulated influx of Ca2+ in non-excitable cells such as epithelial and blood cells. Its contribution to proper phospholipase C–mediated signaling identifies capacitative calcium entry as a major component in the regulation of epithelial cell secretion (discharge of proteins and transepithelial secretion of salts and water) as well as regulation of carbohydrate anabolism and catabolism in the liver. In blood cells, various functions, such as chemotaxis and vesicular secretion of lysosomal enzymes, are subtended by phospholipase C–dependent Ca2+ signaling and capacitative calcium entry (see below). In lymphocytes, capacitative calcium entry cues the mitogenic response to receptor activation, and thus is essential for appropriate clonal expansion. In addition to short-term, receptor-controlled cellular responses, such as secretion, chemotoxis, and muscle contraction, capacitative calcium entry is also involved in longer-term cellular control mechanisms that support cell growth, differentiation, and apoptosis (3).
Noteworthy examples of excitable cells that significantly utilize capacitative calcium entry include smooth muscle cells and endocrine and neuroendocrine cells (14-17). Although capacitative calcium entry may thus play a major role in the functioning of excitable cells, Ca2+ signaling in such cases often involves an “excitation-independent” pathway that is similar to that in non-excitable cells (i.e., via receptor activation of phospholipase C). It has been generally thought that neurons do not exhibit capacitative calcium entry, but a recent study demonstrates that this assumption may be incorrect (18). This issue is especially important, given the recently suggested link between altered capacitative calcium entry and Alzheimer's disease (18, 19).
PHARMACOLOGICAL IMPLICATIONS OF CAPACITATIVE CALCIUM ENTRY
Because of the large number of physiological processes regulated by capacitative calcium entry, specific reagents that modify this signaling process could have clinical utility. At the same time, it might be argued that the ubiquity of capacitative calcium entry would preclude its specific inhibition; inhibitors would probably be nonspecific. Nevertheless, inhibitors of another class of ubiquitous calcium channels, namely, the L-type voltage-dependent calcium channels, have proven extremely useful in treating hypertension and other cardiovascular conditions. Indeed, these inhibitors were sufficiently specific so as to allow for the discovery and understanding of distinct subclasses of voltage-dependent channels. Distinctive selectivities have similarly revealed that multiple kinds of ion channels are involved in capacitative calcium entry (3). As our understanding of the channel subtypes and mechanisms of capacitative calcium signaling is refined, we can expect the development of reagents of increasing pharmacological specificity.
pharmacological activation of capacitative calcium entry
To date, no reagents are known to directly activate capacitative calcium entry. However, there are reagents that will activate this pathway by virtue of their ability to bring about depletion of intracellular Ca2+ stores. In addition to physiological stimuli that generally act through formation of IP3, a number of reagents will cause passive depletion of endoplasmic reticulum stores. Most of these agents act by inhibition of the active uptake of Ca2+ into the endoplasmic reticulum (or, in the case of muscle cells, the sarcoplasmic reticulum). Such agents are known as sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) inhibitors. SERCA inhibitors such as thapsigargin were in fact instrumental in the discovery of the capacitative calcium entry mechanism, and they continue to serve as a useful tools for further study. Ca2+ ionophores, such as A23187 and ionomycin, can also be used to promote the passive depletion of intracellular Ca2+ stores, although they also directly transport Ca2+ across the plasma membrane, potentially confusing the picture of capacitative calcium entry. However, this problem does not arise in electrophysiological studies because the transport of Ca2+ by the ionophores is an electrically silent Ca2+/2H+ exchange. In addition, for reasons that are not entirely clear, ionomycin appears to have some selectivity for endoplasmic reticulum membranes over plasma membranes, especially when employed in the submicromolar concentration range.
Pharmacological Inhibition of Capacitative Calcium Entry
There are a number of drugs that inhibit capacitative calcium entry channels, although in most instances with less than optimal specificity (Table 1⇓). Many of these agents were characterized for inhibition of calcium entry in non-excitable cells years before the conceptualization of capacitative calcium entry. The pharmacologically useful inhibitors of voltage-dependent calcium channels cannot be used at reasonable concentrations to inhibit Ca2+ movements in non-excitable cells.
Inhibitors ofCapacitativeCalciumEntry
The simplest and most dependable of capacitative calcium entry inhibitors are Ca2+ mimics, such as divalent cations, and the potent trivalent lanthanides (20). Lanthanum has a large hydrated ionic radius, which precludes its entry into most cells types (21). Its volume and charge provide high affinity for surface membrane anionic sites, especially those normally occupied by Ca2+ (20). By appropriate adjustment of concentration, lanthanum can thus be used as a relatively specific calcium channel blocker, although at higher concentrations, it also blocks plasma membrane Ca2+ pumping (21, 22).
A more specific, and better-characterized, lanthanide is Gd3+ (Figure 1⇓). Broad et al. demonstrated that in A7r5 smooth muscle cells, capacitative calcium entry could be blocked by as little as 1 μM Gd3+, whereas a parallel entry pathway activated by arachidonic acid was much less sensitive (23). A similar finding was described by Luo et al. in a report comparing capacitative calcium entry and a non-capacitative pathway in HEK293 cells (24). In both studies, the high sensitivity of capacitative calcium entry to Gd3+ provided pharmacological evidence for a receptor-regulated entry mechanism signaled by non-capacitative mechanisms (Figure 2⇓). This high sensitivity to lanthanides is consistent with the finding that capacitative calcium entry channels appear to be the most selective of known Ca2+ channels (at least, in some cell types) (25).
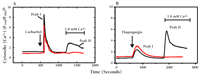
Capacitative calcium entry can be inhibited by gadolinium ions (Gd3+) and activated by thapsigargin.a
A. Cells (in Ca2+-free medium) actively respond to the cholinergic agonist carbachol (100 μM) by releasing internal stores of Ca2+ into the cytosol (Peak I). Subsequently supplied external Ca2+ is taken up by the cells in the absence (black trace; Peak II) but not in the presence (red trace) of Gd3+ (1 μM). Gd3+ is thus an inhibitor of capacitative calcium entry.
B. Thapsigargin (1 μM) causes cells (in Ca2+-free medium) to leak their internal stores of Ca2+ into the cytosol (Peak I). Capacitative calcium entry ensues (black trace; Peak II), which is again inhibited by Gd3+ (red trace).
aData have been simplified from (24).
bF340/F380 is the fluorescence shift of the Ca2+ indicator Fura-2.
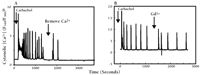
Gadolinium is a specific inhibitor that distinguishes between capacitative and non-capacitative calcium entry.a
A. A single HEK293 cell (in Ca2+-containing medium) responds to the cholinergic agonist carbachol (5 μM) with repetitive spikes in cytosolic Ca2+ concentration; the subsequent removal of Ca2+ from the medium reveals these spikes to be dependent upon Ca2+ influx.
B. A single HEK293 cell (in Ca2+-containing medium) responds to the cholinergic agonist carbachol (5 μM) with repetitive spikes in cytosolic Ca2+ concentration that are not inhibited by Gd3+, thereby indicating that they involve a non-capacitative pathway.
aData have been simplified from (24).
bF340/F380 is the fluorescence shift of Ca2+ indicator Fura-2.
On the other hand, Fernando and Barritt (26, 27) found that submicromolar Gd3+ inhibited divalent cation entry in the presence of vasopressin (i.e., agonist) or thapsigargin (i.e., an activator of capacitative calcium entry) equally; however, in both cases, no more than about 70% of entry could be inhibited. Thus, in the hepatocyte, Gd3+ reveals the presence of both lanthanide-sensitive and lanthanide-insensitive store-operated channels. The latter behavior is reminiscent, in some assays, of TRP channels stably expressed in experimental cells (28).
Because inorganic channel inhibitors tend to engender technical disadvantages, their general use is restricted to simple media and usually to cell culture. Merritt et al. have described an organic inhibitor of receptor-mediated calcium entry in platelets, neutrophils, and endothelial cells (29). Because receptor-mediated Ca2+ entry in these cell types is known to be capacitative, the novel inhibitor, designated SK&F96365, is indicative of antagonism of this pathway. SK&F96365 is specific in blocking the entry phase of the response (i.e., the influx of Ca2+ across the plasma membrane) rather than the release phase (i.e., depletion of Ca2+ from internal stores), and in fact it appeared to be equally potent in blocking voltage-dependent Ca2+ channels. Thus, the compound is likely to be a true channel-blocking substance, because the mechanisms and signaling pathways activating capacitative and voltage-dependent calcium channels are different. Other compounds that block capacitative and voltage-dependent calcium channels with similar potencies include L651582 (discussed below), SC38249 (30), LU52396 (31), and tetrandrine (32); see also (33).
A number of compounds with other well-established biological actions have subsequently been shown also to act as inhibitors of capacitative calcium entry. Notable in this group are the inhibitors of cytochrome P-450, leading to the suggestion that cytochrome P-450 might be involved in the production of a signal for capacitative calcium entry (34). However, like inhibitors discussed above, the P-450 inhibitors do not appear to be particularly selective for capacitative calcium entry as compared to voltage-dependent calcium channels (35). In fact, they are as effective at inhibiting red blood cell potassium channels as they are at inhibiting capacitative calcium entry channels (36).
As pointed out by Clementi and Meldolesi (33), drugs that inhibit enzymes of arachidonic acid metabolism (i.e., cyclooxygenase and lipoxygenase) also tend to inhibit calcium channels. As is true for the P-450 inhibitors, the bases on which inhibitors of arachidonic acid metabolism also inhibit capacitative calcium entry are not clear. The cyclooxygenase inhibitors that inhibit calcium channels include niflumic acid (37), flufenamic acid (38) and tenidap (39, 40), and the lipoxygenase inhibitors with such an activity include nordihydroguaiaretic acid (34, 41) and eicosatetraynoic acid (34, 40, 42). Some of these agents are also P-450 inhibitors, further complicating the mechanistic relationships among the various groups of antagonists. It has been noted that these agents have a variety of non-specific actions that could underlie their inhibitory actions (33), including both the inhibition of mitochondrial ATP synthesis that appears to support capacitative calcium entry (43, 44), as well as disturbances of cytoplasmic pH (45).
An interesting inhibitor of capacitative calcium entry channels that has recently been described is 2-aminoethoxydiphenyl borate (2APB). This membrane-permeant compound was originally shown to be an inhibitor of the IP3 receptor (46). It was subsequently shown to block capacitative calcium entry as well as Ca2+ entry through TRP channels expressed in HEK293 cells (47). Because such TRP channels were not blocked by 2APB when directly activated by diacylglycerols, however, and yet were blocked by 2APB when activated by an agonist coupled to phospholipase C, the IP3 receptor was concluded to promote activation of both TRP and capacitative calcium entry channels. This conclusion has been refined by more recent work suggesting that 2APB acts directly on capacitative calcium entry channels, rather than on intracellular IP3 receptors that activate the channels: First, electrophysiological studies have shown that 2APB acts at the external surface of the cell, rather than intracellularly (48). Second, a cell line shown to be totally lacking in IP3 receptors (due to targeted gene knockout) responds to calcium store depletion with a normal capacitative calcium entry response, and this response is blocked by 2APB (49). 2APB thus appears to be more potent in blocking capacitative calcium entry channels than in blocking IP3 receptors (47, 49, 50). Moreover, it is rather selective for capacitative calcium entry channels, having no effect on voltage-dependent calcium channels ((46) and unpublished observations) or arachidonic acid–gated calcium channels (24) (Figure 3⇓).
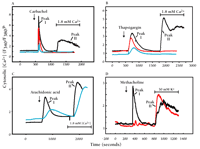
Selective blockade of capacitative calcium entry by 2-aminoethoxydiphenyl borate (2APB).a
A. The response of HEK293 cells (in Ca2+-free medium) to the cholinergic agonist carbachol (100 μM) is shown (black trace) as in Figure 1A. Capacitative calcium entry upon addition of Ca2+ (Peak II) is completely blocked by 30 μM 2APB (red trace). A higher concentration of 2APB (100 μM, blue trace) is required to inhibit the IP3-dependent transient response (Peak I) to carbachol.
B. The response of HEK293 cells (in Ca2+-free medium) to thapsigargin (1 μM) is shown (black trace) as in Figure 1B. 2APB (30 μM, red trace; 100 μΜ, blue trace) also inhibits this response.
C. The activation of channels in HEK293 cells upon treatment with arachidonic acid (30 μM; black trace) is not inhibited by 2APB at 100 μM (blue trace).
D. In AR4-2J cells, a pancreatoma cell line, a phospholipase C–dependent response to cholinergic agonist methacholine (100 μM) as well as L-type Ca2+ channel activation in response to elevated K+ are evident in the absence of 2APB (black trace). As shown by the red trace, 2APB (75 μM) completely blocks the IP3-dependent response, but does not block the activation of L-type voltage dependent Ca2+ channels by elevated external K+.
aData for panels A, B, and C come from (24).
bF340/F380 is the fluorescence shift of the Ca2+ indicator Fura-2.
Recent studies on the signaling mechanisms underlying capacitative calcium entry have revealed reagents that inhibit capacitative calcium entry, but probably not by an action on the channels themselves. Thus, U73122, an inhibitor of phospholipase C, blocks capacitative calcium entry (51), provided that the channels are not already in an activated state (49). In the context of the conformational coupling hypothesis for capacitative calcium entry (see above), U73122 might be taken to indicate a role for IP3 in the signaling mechanism. However, Broad et al. (49) have found that the phospholipase C inhibitor was effective even when the cell was directly supplied with IP3, thereby suggesting a role for phospholipase C in capacitative calcium entry independent of IP3 production. Likewise, a number of laboratories have demonstrated, by use of inhibitors of phosphatidylinositol phosphorylation (such as wortmannin and LY-294002), that polyphosphoinositides are required for activation of capacitative calcium entry (49, 52). As for U73122, these inhibitors do not work once channel activation has taken place (49). And also as for U73122, direct intracellular application of IP3 does not remove the blockade, again indicating a role for polyphosphoinositides in the activation mechanism for capacitative calcium entry that does not involve production of IP3 (49).
CLINICAL USES OF INHIBITORS OF CAPACITATIVE CALCIUM ENTRY
As argued above, the relatively numerous physiological functions of capacitative calcium entry would suggest potential pharmacological strategies for clinical intervention in a number of important diseases. To date, inhibitors of capacitative calcium entry appear likely to be therapeutically useful in two general areas: inflammation and cancer chemotherapy. Although developed as a cyclooxygenase inhibitor, the anti-inflammatory actions of tenidap appear to be due to its ability to block capacitative calcium entry channels; see (33). Thus, Cleveland et al. demonstrated that at clinically useful concentrations, tenidap blocks calcium entry in mast cells activated by antigen as well as by thapsigargin (39). The properties and actions of tenidap are useful in the treatment of rheumatoid arthritis and osteoarthritis (33, 53).
The putative role of capacitative calcium entry in the control of cellular mitogenesis suggests that inhibitors might be useful in cancer chemotherapy. The P-450 inhibitor clotrimazole potently inhibits cellular proliferation, whereas its analog, econazole, is much less effective in this regard (54); this distinction may rest on the ability of clotrimazole, not manifested by other P-450 inhibitors, to inhibit intracellular calcium release as well as calcium entry. These effects occur, with little side effect, in the same concentration range in which clotrimazole is useful as an antimycotic (54). As a result, the drug is currently being investigated clinically as an anti-tumor agent (33).
The potential anti-tumor action of L651582 was disclosed (55) prior to the discovery that the drug appeared to act as an inhibitor of calcium entry. Felder et al. (56) demonstrated that low micromolar concentrations of L651582 appear to specifically inhibit calcium entry without affecting inositol phosphate formation or cAMP formation; slightly higher concentrations (10 μM) inhibit all signaling pathways. The drug is a potent inhibitor of angiogenesis, a process known to be dependent on calcium entry, and through such an action L651582 would be expected to reduce the growth of solid tumors (57). Currently, L651582 is undergoing clinical trials as a treatment for ovarian cancer (55, 58).
PATHOLOGY AND TOXICOLOGY OF CAPACITATIVE CALCIUM ENTRY
There are to date few documented disease states that can be specifically attributed to a malfunction of capacitative calcium entry. However, there is evidence that chemical toxicity and environmental insults may significantly perturb this pathway. The potential for disease associated with capacitative calcium entry should also be obvious, given the central role of calcium influx in myriad physiological systems. Rao et al. documented diminished intracellular Ca2+ signaling, including a decrease in calcium entry, in platelets of certain patients with congenital platelet dysfunction (59). The most specific instance of a disease arising from impaired capacitative calcium entry may be the report of an inherited T-cell immunodeficiency (60) in which a patient displayed a normal number of T-cells that were nevertheless unable to proliferate in response to anti-CD2 or anti-CD3 antibodies; tyrosine phosphorylation and phosphoinositide responses were normal. Release of intracellular Ca2+ by anti-CD3 or by thapsigargin was also normal; however, entry of Ca2+ was defective. Whole-cell patch-clamp recordings showed that the patient's lymphocytes generated no detectable capacitative calcium current upon dialysis of the cell with IP3 or application of thapsigargin or ionomycin (61). This finding emphasizes the essential importance of capacitative calcium entry in the normal functioning of the immune system.
Prolonged elevation of cytoplasmic Ca2+ can be toxic to cells (62, 63); even at moderate levels, Ca2+ can serve as an inducer of cell proliferation or of apoptosis. It is likely, therefore, that agents that augment capacitative calcium entry, whether directly or indirectly, will have untoward effects on cellular physiology. The prototypical example of agents in this class is thapsigargin, which is capable both of killing cells in vitro by apoptosis (64, 65), and of acting as a tumor promoter in vivo (66, 67).
An example of a calcium-mobilizing environmental toxin is tributyltin, an important component of marine paints that has been shown to accumulate in coastal waters and estuaries and in marine organisms. Chow et al. suggest that tributyltin may represent a toxicological mechanism involving capacitative calcium entry (68). Specifically, tributyltin was capable of mobilizing intracellular Ca2+ stores in lymphocytes, thereby activating calcium entry. This action may underlie the known immunotoxicity of tributyltin.
A more widely investigated class of environmental toxins is the polycyclic aromatic hydrocarbons (PAHs). These potentially toxic substances are generated by the burning of fossil fuels, and occur in cigarette smoke and charcoal-cooked food. Chemicals of this class are potent carcinogens and are generally immunosuppressive; it has been suggested that this immunosuppressive action contributes to carcinogenicity (69). Kreiger et al report that PAHs cause a persistent elevation in intracellular [Ca2+] in HPB-ALL T-cells, and that this response correlates well with the immunosuppressive effect of PAHs (70). The same investigators have shown that PAHs are inhibitors of SERCA calcium pumps, indicating a thapsigargin-like action (58). PAHs are known to be complete carcinogens, not requiring the action of a separate promoter; thus, it is possible that the thapsigargin-like action of PAHs provides a tumor promoter activity that culminates in their carcinogenicity.
CONCLUSIONS
Capacitative calcium entry plays a fundamental role in both the immediate and long-term regulation of cells. It follows that unexpected perturbations in this signaling process will have the potential for pathological outcomes, whereas its pharmacological manipulation may find clinical utility in certain disease states. Because the very concept of capacitative calcium entry is rather new, therapeutic approaches to the phenomenon have not as yet been adequately explored or exploited. A summary of potential targets for agents acting on the capacitative calcium entry pathway is illustrated in Figure 4⇓. We may look forward in the future to increasing information on the role of capacitative calcium entry in disease as well as in the therapy of disease.
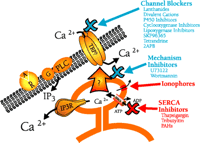
Pharmacology of capacitative calcium entry. Capacitative calcium entry is initiated physiologically by agonists ( ) activating surface membrane receptors (
) activating surface membrane receptors ( ) that are coupled to phospholipase C (PLC), in many instances through a heterotrimeric G-protein (G).Phospholipase C liberates
IP3 from membrane polyphosphoinositides; IP3 in turn binds to its receptor (IP3R) on the endoplasmic reticulum, thereby causing release of stored Ca2+. The fall in stored Ca2+ then, by a mechanism the nature of which is controversial, activates plasma membrane calcium channels. These channels (TRP?)
may be members of the TRP family.
) that are coupled to phospholipase C (PLC), in many instances through a heterotrimeric G-protein (G).Phospholipase C liberates
IP3 from membrane polyphosphoinositides; IP3 in turn binds to its receptor (IP3R) on the endoplasmic reticulum, thereby causing release of stored Ca2+. The fall in stored Ca2+ then, by a mechanism the nature of which is controversial, activates plasma membrane calcium channels. These channels (TRP?)
may be members of the TRP family.
The sites of action (in some instances proposed) for a number of reagents discussed in this review are shown. Those in blue inhibit capacitative calcium entry, whereas those in red activate capacitative calcium entry. The largest group consists of compounds that act, or are presumed to act, as blockers of capacitative calcium entry channels (Channel Blockers). These agents for the most part have limited specificity, with the exception of 2APB, which appears to be specific for capacitative calcium entry channels relative to other known surface membrane calcium channels. U73122 (a phospholipase C inhibitor) and wortmannin (a phosphatidylinositol kinase inhibitor) do not appear to block channels directly, but rather appear to inhibit the mechanism underlying channel activation (Mechanism Inhibitors). Ionophores cause release of stored Ca2+ from the endoplasmic reticulum, and thus bring about activation of capacitative calcium entry. Inhibitors of SERCA pumps, such as thapsigargin or certain toxins, also cause depletion of Ca2+ stores leading to activation of capacitative calcium entry.
- © American Society for Pharmacology and Experimental Theraputics 2001
References

James W. Putney, Jr., Ph.D., is Chief of the Calcium Regulation Section, Laboratory of Signal Transduction, at NIEH-NIH in Research Triangle Park, North Carolina. He is also Adjunct Professor of Pharmacology at The University of North Carolina at Chapel Hill. His interests primarily concern mechanisms of calcium signaling, and specifically mechanisms of regulation of calcium influx across the cell membrane.

