Potassium Channel Subunits: The MiRP Family
- Departments of Pediatrics and Cellular and Molecular Physiology Boyer Center for Molecular Medicine Yale University School of Medicine New Haven, CT 06536
- Address correspondence to SANG. E-mail steve.goldstein{at}yale.edu; fax 203-737-2290.
Abstract
Voltage-gated potassium channels provide tightly Controlled, ion-specific pathways across membranes and are key to the normal function of nerves muscles. They arise from the assembly of four pore-forming proteins called α-subunits. To attain the properties of native currents, α-subunits interact with additional molecules such as the mink-related peptides (MiRPs), single-transmembrane subunits encoded by the KCNE genes. Significantly, mutations in KCNE 1, 2 and 3 have been linked either to life-threatening cardiac arrhythmia or a disorder of skeletal muscle, familial periodic paralysis. The capacity of MiRPs to partner with multiple α-subunits in experimental cells appears to reflect still undiscovered roles for the KCNE-encoded peptides in vivo. Here, we consider these unique peptides in health disease and discuss future research directions.
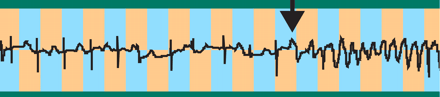
The electrocardiogram shown above demonstrates an arrhythmia, called torsades de pointes (sinusoidal trace at right), that
is incompatible with the normal pump function of the heart. This particular case was drug-induced and reflects the impairment
of specific potassium channels that normally promote the repolarization phase of the cardiac action potential. New insights
into the mutations and drugs that compromise the cellular flux of potassium, not only in the heart but also in other organs,
are being gained through investigations of the protein components of voltage-gated potassium channels.
INTRODUCTION
Potassium channels regulate fluid homeostasis, signaling in nervous tissue, and muscular contraction. This array of tasks is achieved by the controlled and exquisitely timed opening and closing of the water-filled channels through which potassium ions diffuse across cell membranes (1). For example, in the heart, voltage-gated potassium (VGK) channels open in response to a depolarizing wave of excitation that travels through the cells with each beat. The resulting efflux of potassium ions repolarizes the cells, thereby closing the channels and ending the excitation–contraction cycle so that the muscle is prepared to beat again. Similarly, VGK channels in nerve cells facilitate rapid signal transmission and limit excitatory influences. These crucial functions explain why drugs that act on potassium channels, and mutations in channel genes, can have significant effects on human physiology (2).
As shown in Figure 1⇓, each of the four pore-forming α-subunits of VGK channels has six transmembrane (TM) segments and a single pore (P) loop that includes the signature sequence TXGY/FG (3). The tetrameric assemblies form a single, central, ion-selective pore that is lined by the four P loops and the TM segments flanking each P loop in the primary structure (identified as S5 and S6) (4–5). The fourth TM segment of each α-subunit (S4) contains periodic basic residues and acts as the voltage sensor (6–7). In general, the expression of an α-subunit subtype (e.g., KCNQ1, HERG, or Kv3.4) gives rise to functional channels in experimental cells such as Xenopus laevis oocytes or mammalian tissue cells in culture. The currents that arise from these α-subunits, however, are often unlike the native currents manifested by cells in which they are naturally expressed. One well-known explanation for this disparity is the requirement for additional (sometimes called “accessory” or β) subunits—some cytoplasmic, others that are integral to the membrane—that modulate channel properties (8).
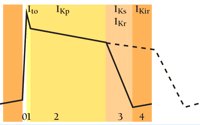
Potassium currents in the cardiac action potential.
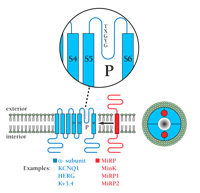
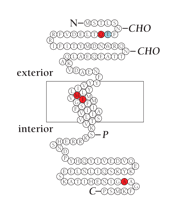
TheKCNE genes encode single-transmembrane–domain peptides (i.e., MiRPs) that regulate voltage-gated potassium (VGK) channels. VGK channels are formed by association of four α-subunits (blue). Each α-subunit contains six transmembrane domains and one pore-forming P loop (P; see inset). P loops contain the pentapeptide sequence TXGY/FG at the ion selectivity filter portion of the pore. In cells, potassium channel α-subunits interact with additional subunits, such as MiRPs (red), as well as membrane and cytosolic regulators (not shown). The MiRPs are encoded by KCNE genes and are a family of β-subunits that assemble (denoted by arrow) with α-subunits to generate native channels. The stoichiometry of MiRP subunits within each channel is not known; a possible α4β2 complex is shown (looking down onto the plane of the exterior membrane surface; right), suggesting close approximation of MiRP and the ion conduction pore (17, 73).
The additionally required subunits that we describe here are encoded by the KCNE gene family and consist of 103 to 177 residues, with a single TM domain. These peptides associate with α-subunits to form stable assemblies with unique properties (Figure 1⇑). The family of peptides encoded by the KCNE genes has five known members, three of which have been characterized. The founding member, MinK, is encoded by KCNE1 and was discovered in 1988 by functional expression of fractionated rat kidney RNA (9). A decade later, KCNE2, KCNE3, and KCNE4 were discovered by focusing on motifs in MinK critical to function using homology-based searches of expressed sequence tag databases; the corresponding gene products were designated the MinK-Related Peptides (MiRPs) 1, 2, and 3 (Figure 2⇓) (8–10). KCNE5, encoding MiRP4, was subsequently found in a gene-rich segment of DNA, the deletion of which results in a syndrome denoted as AMME (for Alport syndrome, mental retardation, midface hypoplasia, and elliptocytosis) (11). MinK has been shown to function in the heart and ear (2), MiRP1 in the heart (10, 12), and MiRP2 in skeletal muscle (13), whereas roles for MiRP3 and MiRP4 are not yet described.

Alignment of amino acid sequences encoded by human KCNE1, 2, 3, 4, and 5. Predicted transmembrane domains are highlighted blue, and highly conserved residues are highlighted yellow. Protein products are named in parentheses next to the respective genes.
MinK, MiRP1, and MiRP2 contribute to the function of VGK channels by influencing channel gating (i.e., channel opening and closing), ion selectivity, and ion flux rate (single channel conductance); they also significantly affect the pharmacology, regulation, and surface expression of VGK channels (10, 12–18). Indeed, concurrent expression of MiRPs and α-subunits (e.g., MinK/KCNQ1, MiRP1/HERG, and MiRP2/Kv3.4) in experimental systems recapitulates the properties of native currents (10, 13, 14), and mutations in KCNE1, KCNE2, and KCNE3 are associated with inherited and acquired disease (10, 12, 13, 19–21). Here, we consider the influence of MinK, MiRP1, and MiRP2 in native potassium channels. Other current reviews have addressed the issues of MinK tissue distribution and regulation (22, 23), structure–function relationships in MinK/KCNQ1 and MiRP1/HERG complexes (8), and the controversial issue of whether the MiRPs act through close juxtaposition with the channel ion conduction pore (24, 25).
MinK, THE KCNE1 GENE PRODUCT
When mRNA that encodes rat MinK is injected into Xenopus laevis oocytes, a very slowly activating potassium current is observed that is similar (but not identical) to the cardiac delayed rectifier current IKs (9, 26). That this 130-residue peptide appeared to form channels on its own produced a controversy only later laid to rest by the cloning of its pore-forming partner, KCNQ1. Specifically, the KCNQ1 gene (originally KvLQT1; first isolated by its linkage to the cardiac rhythm disorder known as long QT syndrome) was recognized to encode a classical VGK α-subunit (27). The paired expression of KCNQ1 with KCNE1 in experimental cells thus reproduced currents that resembled the native IKs of cardiac myocytes (14, 74). MinK-induced currents in oocytes are thus attributable to a Xenopus laevis ortholog of KCNQ1 (14). Indeed, MinK does not generate currents in cells, such as Chinese hamster ovary (CHO) cells, that lack endogenous KCNQ1 (28). In addition to contributing to the formation of IKs channels with KCNQ1, MinK may also influence the function of HERG (see section on MiRP promiscuity, below) (39).
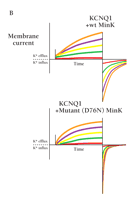
The most apparent effect of MinK on KCNQ1 is an approximately tenfold slowing of activation kinetics (Figure 3⇓), although MinK also alters inactivation and deactivation gating (17, 29). Moreover, the unitary conductance of the MinK/KCNQ1 channels (in symmetric 140 mM potassium ions) is approximately fourfold greater than that of homomeric KCNQ1 channels (17, 30). MinK also alters selectivity for monovalent cations (16, 17), sensitivity to both external and internal pore blockers (18, 31), inhibition by class III anti-arrhythmic agents (i.e., potassium channel blockers) (15), and activation by small-molecule regulators (15).
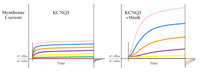
MinK associates with KCNQ1 to generate IKs. The human gene for the α-subunit KCNQ1 can be expressed (here in Xenopus laevis oocytes in whole-cell mode) to generate functional VGK channels (left). Co-expression of MinK is required, however, to generate a current that is like IKs in cardiac myocytes (right). Note that MinK greatly augments KCNQ1-mediated currents, and retards current activation. Cells were held at -80 mV and depolarized for 5 s to voltages of -60 (red), -40 (green), -20 (yellow), 0 (purple), 20 (orange), 40 (blue), and 60 (pink) mV.
Inheritance of single missense mutations in KCNE1 leading to the replacement of serine at position 74 with leucine, or aspartate at position 76 with asparagine (Figure 4A⇓) has been associated with inherited long QT syndrome (17, 19–21, 40). Mutations in KCNE1 reduce net potassium flux by producing a positive shift in the voltage dependence of activation (i.e., less current is generated at a given level of depolarization), a reduction in the single channel conductance, and accelerated deactivation (Figure 4B⇓) (17). Mutations in KCNE1 (and KCNQ1) have been implicated also in predisposition to drug-induced long QT syndrome (50, 51).
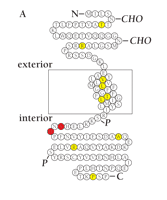
Inherited mutations in the MinK-encoding gene KCNE1 compromise VGK currents and are associated with arrhythmia.
A. Schematic of the MinK peptide indicating disease-associated mutations. Residues highlighted in red represent missense mutations (i.e., S74L and D76N) that have been functionally characterized and clinically associated with arrhythmia (see text for details; also see panel B); residues highlighted in yellow represent mutations that occur in individuals with arrhythmia but that have not been functionally characterized (CHO, predicted sites of glycosylation; P, predicted sites of phosphorylation).
B. Characterization of IKs current arising from mutant MinK. Currents were studied in Xenopus laevis oocytes and measured in excised inside-out patches in symmetrical 100 mM KCl. The KCNQ1 protein was co-expressed either with wild-type MinK or mutant form of MinK that is associated with Jervell and Lange-Nielsen syndrome. Patches were held at -60 mV and depolarized for 6 s to voltages of 0 (red), 20 (green), 40 (yellow), 60 (purple), and 80 (orange) mV. (NB: The reduction in single channel conductance associated with the mutant MinK is not indicated; see text for details.)
Individuals who carry two mutant alleles of KCNE1 can suffer from a prolonged QT interval combined with sensorineural deafness (i.e., Jervell and Lange-Nielsen syndrome; JLNS) (21, 40). Congenital deafness in these cases stems from disrupted production of endolymph due to defective IKs channels in the stria vascularis in the cochlea (41). Thus, a point mutation in KCNE1 has pathological consequences in multiple tissues, indicative of a channel complex with multiple roles. It should be noted that KCNE1 transcripts are found in heart, auditory epithelium, kidney, eye, gastrointestinal tract, and T cells (22, 23).
MiRP1, THE KCNE2 GENE PRODUCT
Even as HERG was becoming recognized as the likely molecular correlate for IKr based on transcript levels and function, its failure to fully recapitulate native IKr currents on its own led to the suggestion that an additional subunit might be required (43). Study of KCNE2 resolved the disparity between native and cloned (i.e., homomeric) HERG channels; MiRP1 and HERG were found to form stable complexes that behave like native IKr channels (Figure 5⇓) in terms of their unitary conductance, sensitivity to external potassium ions, deactivation rate and, notably, inhibition by the anti-arrhythmic agent E-4031; thus, homomeric HERG channels were inhibited by E-4031 only in a use-dependent fashion during repetitive pulse cycles, whereas MiRP1/HERG and native IKr channels show tonic block (prior to the first activating pulse) and ready relaxation to equilibrium blockade. Although unitary conductance and gating kinetics were altered by MiRP1, the distinctive gating attributes of HERG-containing channels were qualitatively unchanged: With depolarization, the channels activate but generate little outward current due to rapid inactivation; upon repolarization, transition to the closed state is slow, allowing for further ion flux (42).
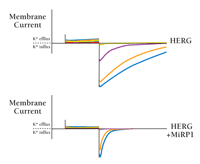
MiRP1 associates with HERG to generate IKr. The human gene for the α-subunit HERG can be expressed (here in Xenopus laevis oocytes in whole-cell mode) to generate functional VGK channels. Co-expression of MiRP1 is required, however, to generate a current that is typical of IKr in cardiac myocytes. Note that MiRP1 greatly decreases HERG-mediated currents and accelerates channel deactivation. Cells were held at -80 mV and depolarized for 3 s to voltages of -60 (red), -40 (green), -20 (yellow), 0 (purple), 20 (orange), and 40 (blue) mV, and were subsequently repolarized to –100 mV.
Consistent with a role in repolarization of the myocardium, mutations that impair MiRP1 function are associated with inherited cardiac arrhythmia (10). Four mutations in KCNE2 have been found that correlate to a prolonged QT interval and/or TdP (Figure 6⇓): the Q9E substitution (glutamine to glutamate at position 9) within the predicted extracellular portion of MiRP1; M54T (methionine to threonine) and I57T (isoleucine to threonine) substitutions in the predicted TM domain; and A116V (alanine to valine) in the predicted cytoplasmic domain. All four mutations diminish baseline potassium flux through the MiRP1/HERG channels: the Q9E substitution shifts the voltage dependence of activation to more positive values; the M54T substitution accelerates channel deactivation; and the I57T and A116V substitutions apparently compromise unitary current magnitude and cell surface half-life, respectively (F. Sesti, personal communication).
Mutations of the MiRP1-encoding geneKCNE2 can severely compromise VGK currents and are associated with arrhythmia. The schematic of MiRP1 indicates disease-associated mutations. Residues highlighted in red represent missense mutations (i.e., Q9E, M54T, I57T, and A116V) that have been functionally characterized and clinically associated with arrhythmia (see text for details); the residue highlighted in blue represents the site of a polymorphism (i.e., T8A) associated with drug-induced arrhythmia (CHO, predicted sites of glycosylation; P, predicted sites of phosphorylation).
Although inherited arrhythmia is rare, acquired arrhythmia is relatively common in association with adverse reactions to medications, drugs of abuse, and metabolic abnormalities. As is true of most common disorders, acquired long QT syndrome appears to have a multifactorial basis. Thus, patients often present with multiple inciting factors (e.g., hypokalemia and female sex) that act cumulatively to impede cardiac repolarization (37, 44). Of particular clinical relevance is the increased likelihood that individuals with a prolonged QT interval may develop arrhythmia in response to drug intake (45, 46). Thus, individuals harboring the M54T, I57T, and A116V variants of MiRP1 came to clinical attention when they ingested procainamide, oxatomide, and quinidine, respectively, three agents that inhibit IKr channels and delay myocardial repolarization (12). In these cases, the corresponding KCNE2 mutations do not alter sensitivity of the channel to the given drug. Conversely, the Q9E variant of MiRP1, identified in a patient with a long QT interval at baseline in whom clarithromycin had induced TdP and VF, not only affects channel gating to reduce potassium flux, but increases sensitivity of MiRP1/HERG channels to inhibition by the drug (10), a macrolide antibiotic previously associated with long QT syndrome and IKr blockade (47). The patient, moreover, was female and hypokalemic when she developed arrhythmia, two other factors associated with reduced IKr current density. The consequences of KCNE2 mutations thus underscore the need to consider acquired long QT syndrome in multifactorial terms.
In contrast to the patients characterized by rare (less than 0.02 percent of the general population) mutations of channel genes such as KCNE2, many more individuals appear free of an “inherent” risk of arrhythmia (i.e., their electrocardiogram is normal) but are nevertheless prone to acquire arrhythmia in response to certain drugs. In particular, the T8A polymorphism of MiRP1 is present in ~1.6 percent of the general population, and although it is associated with neither inherited long QT syndrome nor changes in MiRP1/HERG channel function (10), the variant increases sensitivity to blockade by a common antibiotic (leading to 50% current suppression at levels of the drug present in serum) and is implicated in drug-induced arrhythmia (12). Thus, a common polymorphism can carry important clinical implications for choice of drug therapy where alternatives exist, as well as for future drug development.
Because IKr channels appear to be particularly susceptible to blockade by a range of drugs, including anti-arrhythmic agents, antibiotics, antihistamines, and psychotropic drugs (48), it has been proposed that several unique structural features of HERG make it prone to drug blockade. These include a large internal channel cavity bearing aromatic residues that may facilitate binding and trapping of small molecules (49). The structural characteristics of MiRP1 that contribute to the susceptibility of IKr to drug blockade have not yet been elaborated.
MiRP2, THE KCNE3 GENE PRODUCT
MiRP2 is only 103 residues long, with a short predicted intracellular domain (Figure 7⇓) (8, 10, 13). In contrast to KCNE1 and KCNE2, transcripts of the KCNE3 gene (encoding MiRP2) are most abundant in skeletal, rather than cardiac, muscle. The search for a pore-forming α-subunit that might partner with MiRP2 to form native potassium currents led to Kv3.4, a VGK channel protein prominent in skeletal muscle. Kv3.4 was found to be coordinately expressed with MiRP2 in rat sartorius and murine C2C12 cell plasma membranes and to form stable assemblies with MiRP2 that altered the biophysical and pharmacological properties of Kv3.4 to match those of a native muscle channel (13).
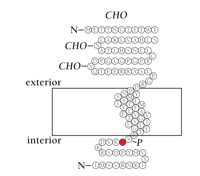
Schematic of the MiRP2 peptide indicating a mutation associated with familial periodic paralysis. The residue highlighted in red represents a missense mutation (i.e., R83H) that is associated with disease (CHO, predicted sites of glycosylation; P, predicted sites of phosphorylation).
Related to the Drosophila Shaw family of potassium channels, homomeric Kv3.4 channels activate at supra-threshold potentials and inactivate rapidly (i.e., A-type behavior). These properties, however, are not observed in skeletal muscle, although Kv3.4 protein is conspicuous. Dependence of the native current on association with MiRP2 seems to explain this finding, at least in part, as MiRP2/Kv3.4 channels activate at sub-threshold potentials (with a midpoint voltage dependence ~45 mV more negative than homomeric Kv3.4 channels), and like native channels in C2C12 skeletal muscle cells, their steady-state activity at negative voltages is ~80-fold greater than channels that lack MiRP2 (Figure 8⇓) (13). MiRP2 also decreases channel sensitivity to the sea anemone toxin BDS-II, producing an inhibition constant similar to that manifested by native channels in muscle cells, and endows Kv3.4 subunits with a single channel conductance and rate of recovery from inactivation typical for native channels (13).
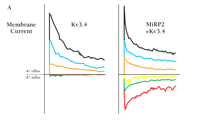

The KCNE3-encoded peptide MiRP2 associates with the α-subunit Kv3.4 to generate VGK channel currents typical of skeletal myocytes.
A. The human gene for the α-subunit Kv3.4 can be expressed in CHO cells to generate functional VGK channels. Co-expression of MiRP2 is required, however, to generate a current that is typical of skeletal myocytes (right); note that MiRP2 leads to sub-threshold currents. Cells were held at -80 mV and depolarized for 3 s to voltages of -70 (red), -40 (green), -20 (yellow), 20 (purple), 40 (orange), 70 (blue), and 90 (black) mV. Kv3.4 currents were studied in on-cell patches with 100 mM external KCl.
B. Steady-state currents in patches held at –40 mV. The displayed traces represent 18-second continuous recordings at 6 s per line. (The patches representing expression of Kv3.4 and co-expression of Kv3.4/MiRP2 in CHO cells contain one channel; native C2C12 myocyte patches contain three channels.)
The observed effects of MiRP2 alter our expectations for the role for Kv3.4 subunits in skeletal muscle physiology. Previously, Kv3.4 channels were hypothesized to modulate calcium currents (owing to their supra-threshold activation) and to produce afterhyperpolarization (due to slow recovery from inactivation), thereby limiting action potential frequency (52). In contrast, MiRP2/Kv3.4 channels activate at sub-threshold potentials and, thereby, contribute to the skeletal muscle resting membrane potential (13). Moreover, their rapid recovery from inactivation suppresses afterhyperpolarization and minimizes cumulative inactivation to facilitate rapid trains of action potentials (Figure 9⇓).
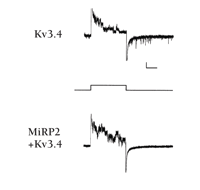
MiRP2 accelerates inactivation recovery of Kv3.4 channels. Representative low current density on membrane patches from CHO cells expressing Kv3.4 or MiRP2/Kv3.4 cannels during a train of 1-s repetitive pulses to +60 mV from a holding voltage of –80 mV (protocol in inset). Wherease Kv3.4 channels show re-openings at –80 mV following depolarization as they recover slowly from inactivation, MiRP2/Kv3.4 channels recover more quickly and re-openings are not visible.
As in the case of KCNE1 (MinK) and KCNE2 (MiRP1), mutation of the KCNE3 gene has been associated with compromised muscle function. The familial periodic paralyses are disorders of skeletal muscle that had previously been associated with mutations in the SCN4A sodium channel (53-55) and in the CACNL1A3 L-type calcium channel (56). Based on identification of MiRP2 and its role in a prominent skeletal muscle current, 100 patients with periodic paralysis but no mutations in genes previously associated with the disease were screened for alterations in KCNE3. Two pedigrees with a missense mutation (producing an arginine to histidine substitution, R83H, in MiRP2) were thereby identified; the mutation was found only in family members with symptoms, but not in unaffected relatives or control individuals, and resulted in diminished potassium flux, reducing the capacity of the channels to pass current and set resting membrane potential (13).
Although KCNE3 is the first potassium channel gene associated with inherited periodic paralysis, the effects of R83H MiRP2 resemble those of mutations in sodium and calcium channels: membrane depolarization, compromised repolarization, and diminished excitability (57). These same effects are seen in acute periodic paralysis caused by barium toxicity, which is thought to result from decreased potassium flux due to channel blockade (58). Although periodic paralyses have tended to be grouped together according to clinical findings, the disorders are quite heterogeneous, with overlapping signs, symptoms, and responses to therapy; gene-based classification is now beginning to aid in both diagnosis and treatment strategies (59, 60).
MiRP1 PROMISCUITY
The MiRPs have been demonstrated to interact with multiple α-subunit partners in experimental cells. Whether this promiscuity indicates a versatility of MiRP function in the physiology of potassium channels remains an open question. Whereas MinK assembles with KCNQ1 to form cardiac IKs currents (14), MinK antisense oligonucleotides reduced IKr currents in murine atrial tumor cells (61), thereby suggesting an interaction between MinK and HERG. Indeed, MinK was found to form stable complexes with HERG in CHO and COS cells and to double current density, apparently by increasing the fraction of active channels in the membrane (39). Consistent with these findings, IKr currents were diminished in cardiac muscle cells from mice where Kcne1 was replaced by lacZ (62). Nevertheless, MinK/HERG complexes do not recapitulate the classical properties of IKr as do MiRP1/HERG complexes, and HERG binds MiRP1 in preference to MinK (10). Conceivably, MinK may assemble with HERG in cells in which MiRP1 is not produced, or may influence MiRP1/HERG complexes to alter their regulation, trafficking, or activity levels.
MiRP1 also exhibits promiscuous interactions. In addition to assembling with HERG to form cardiac IKr channels (10), it associates with Kv4.2 subunits (which contribute to cardiac and neuronal transient outward currents, Ito) to slow channel inactivation and alter the kinetics of 4-aminopyridine blockade (63, 64). Again, it is unclear whether MiRP1/Kv4.2 complexes form in native cells. MiRP1 has also been reported to alter the properties of KCNQ1 when the two subunits are expressed in COS cells (65), although no such effect was seen in two studies using Xenopus laevis oocytes (10, 66) and one using CHO cells (F. Sesti, personal communication).
MiRP2 appears to enjoy a particularly wide range of interactions. In addition to its assembly with Kv3.4 in native and experimental cells (13), MiRP2 can be expressed along with KCNQ1 in CHO cells to produce a channel that is open at resting membrane potentials, and in oocytes MiRP2 suppresses currents passed by HERG and KCNQ4 α-subunits (67, 68).
Moreover, equivalent positions in MinK and MiRP2 appear to act similarly with their recognized pore-forming partners. Thus, D76N MinK (associated with long QT syndrome) alters KCNQ1 function by shifting activation to positive potentials, reducing single-channel current amplitude, and lowering steady-state open probability; D90N MiRP2 (the analogous mutation) affects Kv3.4 and KCNQ1 in the same ways (17, 69). Similarly, R83H MiRP2 (associated with periodic paralysis) shifts activation to positive potentials, reduces unitary conductance, and lowers steady-state open probability of complexes with Kv3.4 and KCNQ1, and the equivalent mutation in MinK (K69H) has similar effects on KCNQ1 channels (69). Remarkably, studies with MinK/MiRP2 chimeras and KCNQ1 have revealed that a three-residue stretch in the TM domain (amino acids 57–59 in MinK, 71–73 in MiRP2) is necessary and sufficient to account for contrasting effects of the two peptides on channel activation (68). Although these results suggest that each MiRP combines with different pore-forming partners to produce similar effects, MiRP2 suppresses HERG currents yet increases flux through KCNQ1 and Kv3.4 channels (13, 67).
CONCLUSIONS AND FUTURE DIRECTIONS
The clinical classification, prognosis, and treatment for patients with ion channel defects are often challenging due to overlapping signs and symptoms. Nevertheless, the clear correlations among mutations of KCNE genes, patient presentation, and disruption of potassium currents has identified important roles for MiRPs in normal physiology and disease. Thus, patients with long QT syndrome can now be assessed in terms of genotype, in addition to clinical presentation and response to drug therapy, and treatment of patients with periodic paralysis has benefited from the use of genotype as a criterion for therapeutic approach (70, 60).
The multifactorial basis of drug-induced arrhythmia has made the identification of patients at risk for adverse drug reactions rather difficult. Gene-based diagnosis now presents itself as a viable option. Indeed, as the database of allelic variants grows, preprescription genotyping will become a consideration for avoiding drug-induced pathology. Moreover, because MiRP peptides alter the pharmacology of cloned VGK channel α-subunits to recapitulate currents typical of native cells, drug development programs may benefit from study of heteromeric complexes that include both a MiRP and its cognate α-subunit.
The essential role of MiRPs in native channels is evident in congenital disorders arising from KCNE mutations. Nevertheless, it is important to note that animal models and human physiology often differ substantially. Kcne1 knockout (-/-) mice, for example, display loss of MinK, disrupted potassium secretion, degeneration of the inner ear, and profound deafness (41, 71), accurately modeling in these respects patients with JLNS; the mice, however, have only minor changes in cardiovascular function (62, 72). Thus, action potentials from Kcne1 (-/-) knockout mice remain normal, whereas long QT syndrome and life-threatening arrhythmia are associated with KCNE1 missense mutations in humans. Similarly, disruption of KCNQ1 in mice (encoding the partner for MinK in IKs formation) results in vestibular deafness but not in electrocardiographic abnormalities (47), although a subsequent report describes conditions similar to JLNS, with both inner ear and cardiac dysfunction (75). Interspecies differences have been attributed to low levels of MinK in adult mice and to the relatively minor role for IKr and IKs in cardiac repolarization in newborn animals (62).
Thus far, MinK/KCNQ1, MiRP1/HERG and MiRP2/Kv3.4 are the recognized MiRP/α-subunit partnerships that have been correlated to native currents and disease. However, as MinK and MiRP2 interact with KCNQ1, and MinK, MiRP1, and MiRP2 all interact with HERG, and all three MiRPs enjoy widespread tissue expression, these recognized partnerships may be the tip of the iceberg. Indeed, there is no reason to presume that MiRP interactions are precluded with calcium-activated potassium channels, cyclic–nucleotide gated channels, inward rectifier potassium channels, and even two-P-domain potassium channels. Among many questions that remain to be answered are the rules that govern subunit specificity and whether complexes can contain more than one type of MiRP. As evidenced by their role in health and disease, these diminutive subunits appear to be more powerful than might be expected based on their size.
Potassium currents of four types contribute to the cardiac action potential: transient outward (Ito), leak (IKp), delayed rectifiers (IKr and IKs), and inward rectifier (IKir); see (32, 33). Calcium and sodium channels are key to phase 0 depolarization (i.e., a positive shift in the cell voltage from resting membrane potential). Ito currents are activated by this depolarization to produce phase 1 repolarization, a partial return toward resting potential as potassium leaves the cell through channels, putatively formed by Kv1.4, Kv4.2 and/or Kv4.3 subunits, that inactivate rapidly. The plateau of the action potential (phase 2) is maintained by calcium and potassium currents (variously called IKp, IKsus, or IKur) that appear to be carried by Kv1.5 and/or KCNK-type 2–P-domain leak channels (34). IKr and IKs, the delayed rectifiers, are activated more slowly upon depolarization and return cells to rest during phase 3 repolarization; IKr and IKs appear to be carried by MiRP1/HERG and MinK/KCNQ1 channels, respectively, as we discuss here. The rise from rest to threshold (phase 4) determines heart rate and is mediated by inwardly rectifying potassium currents (IKir) including IK1 (generated by Kir2.1-4 subunits), IKAch (attributed to Kir3.1 and Kir3.4 subunits) and IKATP (arising from Kir6.1 and SUR subunits) as well as a non-selective cation current (IF) formed of HCN family subunits.
The majority of mutations and polymorphisms that have been associated with arrhythmia occur in genes for the delayed rectifier channels IKr and IKs. In these cases, arrhythmia results from reduction of potassium flux, impairing repolarization capacity, and prolonging the cardiac action potential (see dashed line in Figure⇑). This prolongation is reflected on the surface electrocardiogram as a long QT interval and predisposes patients to torsades de pointes* (TdP), ventricular fibrillation (VF), and sudden death (35–37). Reduced flux can result from channels with suboptimal gating kinetics (e.g., slow rates of opening or passing ions, or increased rates of closing, perhaps effected by drugs); reduced flux can alternatively result from a reduced number of channels in the plasma membrane. Of the six loci that have been associated with long QT intervals, two have been identified as KCNE genes (KCNE1 and KCNE2); two others have been ascribed to genes for the respective α-subunit partners (KCNQ1 and HERG); one is linked to a sodium channel gene (SCN5A); and the final locus has not been correlated to a specific gene (38).
- © American Society for Pharmacology and Experimental Theraputics 2001
References

Geoffrey W. Abbott, Ph.D., is a Postdoctoral Associate in the Section of Developmental Biology and Biophysics and Boyer Center for Molecular Medicine, Yale University. He will soon move to Weill Medical College of Cornell University, Division of Cardiology, as an Assistant Professor of Medicine and Pharmacology.
Steve A. N. Goldstein, M.D., Ph.D., Professor of Pediatrics and Cellular and Molecular Physiology, is Chief of the Section of Developmental Biology and Biophysics and Faculty Member of the Boyer Center of Molecular Medicine, Yale University. His rabid devotion to ion channels was piqued during postgraduate training with Chris Miller at Brandeis University (1989–1993). He is the recipient of the E. Mead Johnson Award (2001).

