The Obesity Epidemic: Pharmacological Challenges
- Stephen R. Bloom1,
- Francis P. Kuhajda2,
- Ismail Laher3,
- Xavier Pi-Sunyer4,
- Gabriele V. Ronnett5,
- Tricia M.M. Tan1 and
- David S. Weigle6
- 1Hammersmith Hospital, Imperial College , London, UK W120NN;
- 2Departments of Pathology, Oncology, and Biological Chemistry, and
- 3Departments of Neuroscience, Neurology, The Johns Hopkins University School of Medicine, Baltimore, MD 21205;
- 4Faculty of Medicine, University of British Columbia, Vancouver, BC, Canada;
- 5St. Luke’s/Roosevelt Hospital, Obesity Research Center, New York, NY 10025;
- 6University of Washington School of Medicine, Harborview Medical Center, Seattle, WA 98104
Abstract
Obesity, defined by a body mass index greater than 30kg/m2, claims an increasing number of lives every year, underscoring a dire need for effective therapeutic interventions. The origins of the obesity epidemic are complex, but commonly cited factors include the large quantities of calorie-rich food that are readily accessible in modern society; eating habits adapted to fast-paced lifestyles; low levels of physical activity; and genetic programs that have evolved, especially in populations prone to famine, to favor the storage of excess calories (i.e., the thrifty-gene theory). It is estimated that more than thirty percent of adults, and about fifteen percent of juveniles, are obese. These high rates have led to dramatic increases in diseases such as type 2 diabetes, cardiovascular and respiratory diseases, depression, and some forms of cancer.

The Global Burden of Obesity1
The International Obesity Taskforce estimates that nearly 1.7 billion people in the world may be at risk for weight-related disease, and current mortality rates (more than 2.5 million deaths per year) due to high body mass index (BMI) are expected to double by the year 2030 [see (1) and (2)]. There are regional and intraregional differences when overweight and obesity are considered on a global perspective. For example, average BMI levels in Africa and Asia are 20–23 kg/m2, whereas a higher average range (25–27 kg/m2) prevails in Europe and North America. On the African continent, BMI levels reach as high as 22 kg/m2 for males and 27.1 kg/m2 for females in South Africa, but even within this country, ethnic origins and gender can influence body weights: Rates of obesity (i.e., BMI greater than 30) are eight percent among black men; twenty percent among white men; thirty percent among black women; and twenty percent among Indian and Asian ethnic groups. In some northern African countries, the picture in terms of percentages can look similar: Half of women in Tunisia are overweight (i.e., BMI between twenty-five and thirty) or obese, a threefold increase over the last twenty years. A threefold increase in obesity in twenty years’ time (1980–2001) has also occurred in England, and even more troublesome, the rate of morbid obesity (defined by a BMI greater than forty) tripled among English men and doubled among English women in the 1990s. More generally, it has become rare to record obesity rates below ten percent in Europe. In the USA, obesity affects a third of all adults and as much as forty to fifty percent of certain populations, such as Mexican American and African American women, with as much as a fifteen-percent incidence of morbid obesity among the latter. Although definitions of obesity have varied elsewhere in the world, such as China, Figure 1⇓ summarizes obesity rates from several regions. The global impact of obesity could also be described in economic terms: Two to seven percent of total health care costs in the US and UK are attributed to weight-related disease; in Tonga, costs related to obesity and type 2 diabetes consume sixty percent of the health care budget.
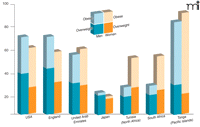
Prevalence of obesity in men and women among selected populations around the world. Rates of overweight (i.e., BMI between twenty-five and thirty) and obesity (i.e., BMI above thirty) can vary significantly within geographical regions according to several factors (e.g., ethnicity; see text for details). [Data from the International Obesity Taskforce (1, 2). Data not standardized according to time period of collection or age ranges included.]
The Early Years of Anti-Obesity Pharmacotherapy: Lessons Learned
The global epidemic of obesity is attended by enormous human and economic costs. Strategies to combat overweight and obesity (Table 1⇓) have been deceptively simplified into a single logical imperative for the patient—energy expenditure should exceed energy storage (Figure 2⇓). This thermodynamic principle guided efforts to develop anti-obesity pharmacotherapy prior to current clinical appreciation for physiological regulators of appetite and energy balance. Thus, the first drugs to be used as anti-obesity agents were chosen, often with disastrous consequences, to promote anorexia (e.g., amphetamines and digitalis) or thermogenesis (e.g., thyroxine and dinitrophenol) (3). Dinitrophenol, which increases metabolic rate by uncoupling mitochondrial oxidative phosphorylation, was introduced in 1933. This agent caused rapid weight loss but was associated with sensations of warmth, sweating, and elevated body temperature. Its use was soon discontinued, owing to severe liver, bone marrow, skin, and retinal toxicity, along with an increase in mortality. Thyroxine, used as a single agent, was also successful in causing weight loss by accelerating metabolic rate, but only at the expense of significant adrenergic side effects and myocardial toxicity. In 1938, amphetamine was found to induce both anorexia and increased sympathetic activity, resulting in significant weight loss. The infamous “Rainbow Pills,” consisting of amphetamine, thyroxine, digitalis, and a diuretic, were used to treat obesity from the 1940s through the 1960s, when they were finally acknowledged to have severe addictive potential and induce sudden cardiac death. In 1965, an anorectic amphetamine derivative called aminorex was introduced in Europe, but within three years it was associated with pulmonary hypertension and a high mortality rate, prompting its market withdrawal. In 1992, the anorectic serotoninergic drug fenfluramine was found to produce significant and sustained weight loss when used in combination with the sympathomimetic agent phentermine. Millions of prescriptions for “fen-phen” were written over the ensuing five years, but pulmonary hypertension, reminiscent of the aminorex experience, and valvular heart disease caused both fenfluramine and dexfenfluramine to be removed from the market in 1997. This long list of anti-obesity drug failures highlights our need to better understand the energy homeostatic system in order to develop effective therapies.
Theoretical Methods for Treating Obesity
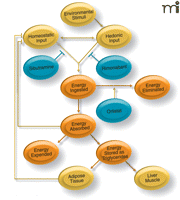
Energy homeostasis and the action of currently available anti-obesity drugs sibutramine, rimonabant, and orlistat. Orange arrows indicate pathways of energy flow. Yellow arrows indicate positive (with arrowheads) and negative (blunt arrows) regulatory pathways. Wholesale environmental change by a societal approach is theoretically preferable but so far seems impracticable. Presently licensed medical treatments such as orlistat, acting in the gut to impair fat absorption, and sibutramine and rimonabant, acting in the CNS to impair appetite, only cause modest loss of weight, which is not sustained once treatment is discontinued. Moreover, there are significant side effects, which may limit their applicability. The only effective long-term intervention at present is bypass bariatric surgery, but this is expensive, incurs a significant morbidity and mortality, and deliverability is limited by the availability of suitable surgeons and support services. New pharmacological approaches to the treatment of obesity are therefore desperately needed.
Central Circuits and Peripheral Signals in Energy Balance
Energy balance depends on the concerted regulation of two principal systems. On the one hand, central nervous system (CNS) circuits must coordinate and integrate appetite, food-seeking behavior, thermoregulation, and the hedonic aspects of eating. On the other hand, peripheral signals of satiety (e.g., from the gut) and overall energy balance must be registered and fed back to the CNS. The brainstem and the hypothalamus are crucial to CNS circuits that regulate appetite. The brainstem receives signals from the periphery via the area postrema, where the blood brain barrier is not complete, and the vagus nerve. The dorsal vagal complex integrates these signals and projects to the hypothalamus, which in turn is also influenced by higher brain centers. The proximity of the hypothalamus to the median eminence, a region that lacks a complete blood brain barrier, may also expose hypothalamic neurons to circulating factors.
Two populations of neurons, which have opposing effects on food intake, reside in the arcuate nucleus of the hypothalamus (Figure 3⇓). The first of these, the medial arcuate nucleus, expresses neuropeptide Y (NPY) and agouti related peptide (AgRP), both of which are orexigenic (i.e., stimulate food intake and food-seeking behavior). In contrast to these orexigenic effects, the second population of neurons, in the lateral arcuate nucleus, expresses pro-opiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART), both of which inhibit food intake. The balance of activity between these two groups of neurons therefore regulates body weight, via the paraventricular nucleus, and engages higher brain centers that regulate 1) food-seeking behavior; 2) basal metabolic rate (via the thyroid axis); and 3) the sympathetic nervous system.
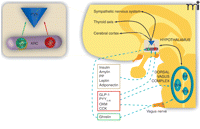
Integration of peripheral signals and central neuronal mechanisms in energy homeostasis and body weight. The hypothalamus and the dorsal vagal complex in the brainstem are central to the regulation of appetite. Hypothalamic circuits that control appetite are schematized in the inset (ARC, arcuate nucleus; PVN, paraventricular nucleus; NPY, neuropeptide Y; AgRP, Agouti related peptide; POMC, pro-opiomelanocortin; CART, cocaine and amphetamine regulated transcript). NPY- and AgRP-expressing neurons stimulate appetite (i.e., are orexigenic; green color); POMC- and CART-expressing neurons counterbalance this by reducing appetite (i.e., are anorexigenic; red color). The dorsal vagal complex comprises three nuclei: the area postrema (AP), the dorsal vagal nucleus (DVN), and the nucleus tractus solitarius (NTS). Peripheral signals are shown in boxes at the left (PP, pancreatic polypeptide; PYY3-36, peptide YY3-36; OXM, oxyntomodulin; CCK, cholecystokinin). The dashed arrows indicate their probable sites of action on the central mechanisms.
Peripheral signals, originating from the gut and adipose tissue, can be divided into two general categories. Short-term signals arise upon the ingestion of a meal to indicate postprandial satiation and terminate eating. Long-term signals communicate the overall level of energy stored in the body, mainly in the form of adipose tissue. Both categories of peripheral signaling represent opportunities for pharmacological intervention into appetite and body weight. The following discussion will consider each signal in turn and current trends in terms of drug development.
Peptide Hormones as Pharmacotherapeutic Targets
Ghrelin
Ghrelin was originally characterized as the endogenous ligand for the growth hormone secretagogue receptor GHS-R1a. It is a twenty-eight–residue peptide with an octanoyl group attached to the serine side chain at position 3. Octanoylation is essential to the agonist activity of ghrelin at GHS-R1a. By far, the richest source of ghrelin in the body is the stomach, although ghrelin is also found in the pituitary and elsewhere (4, 5).
Apart from its growth hormone–releasing actions on somatotroph cells in the pituitary, ghrelin is a powerful stimulator of appetite. It increases food intake, body weight, and adiposity when administered to rats either peripherally or intracerebroventricularly (6, 7). When administered intravenously to healthy human volunteers at a free-choice buffet, ghrelin increases appetite and food intake thirty percent (8), and similar effects are seen with subcutaneous injections (9). Ghrelin levels naturally rise upon fasting, and fall after eating, consonant with a “hunger hormone” role (10).
In addition to its short-term effects on hunger, ghrelin is also thought to signal longer-term changes in energy balance. Ghrelin levels are negatively correlated with BMI and rise after weight loss induced by diet or bariatric surgery (11). The exception to this pattern is in patients with Prader-Willi syndrome, who despite high adiposity have elevated circulating ghrelin levels, which is perhaps the basis for symptomatic hyperphagia (12). Similarly, postprandial reduction in ghrelin concentration is not observed in some obese individuals, suggesting that sustained levels of ghrelin may contribute to obesity (13, 14). Resistance to ghrelin does not appear to typify obese subjects, as ghrelin infusions stimulate appetite and food intake regardless of body weight (15).
GHS-R1a Antagonists
The GHS-R1a antagonist [D-Lys-3]-GHRP-6 has been shown to reduce food intake in lean, diet-induced obese, and ob/ob mice (16); two antagonists in development are BIM-281163 (Ipsen Group) and JMV 2760 (Æterna Zentaris Inc.). JMV 2760 apparently elicits an eighty percent–reduction in food intake stimulated by the GHS-R1a agonist hexarelin (17). The Ipsen compound has mixed effects, however, as it blocks ghrelin-induced GH release, as expected, but also induces long-term weight gain (18). This counterintuitive effect may be mediated through an unknown alternative GHS receptor. GHS-R1a has a high constitutive activity (19), which suggests that an inverse agonist might be advantageous in reducing the activity of GHS-R1a. Intriguingly, a substance P–based compound, [(D-Arg1, D-Phe5, D-Trp7,9, Leu11)-substance P) has such inverse agonist activity (20), although its in vivo effects are not reported.
NOXXON Pharma has recently developed an oligonucleotide-based compound called an RNA Spiegelmer, similar in principle to RNA aptamers but employing L-ribose in the sugar-phosphate backbone instead of the natural D-ribose. Spiegelmers are resistant to nuclease activity and can persist in the circulation for up to sixty hours, an effect sustained by coupling them to polyethylene glycol (PEG). The lead compound L-NOX-B11 was isolated by exponential enrichment, a PCR-based technique that selects for oligonucleotides binding a target, in this case ghrelin. L-NOX-B11, which binds to the N-terminal pentapeptide of ghrelin, including the vital octanoyl group, blocks the stimulation of GH release and food intake in mice (21, 22). A second-generation Spiegelmer, PEGylated L-NOX-B11-2, induces weight loss in a mouse model of diet-induced obesity (23).
Immunization against Ghrelin
In mice, immunization against ghrelin has been shown to prevent weight gain without altering food consumption (24). However, a double-blind phase I/IIa trial of the vaccine CYT009-GhrQb in 111 obese individuals showed no effect on body weight despite a good antibody response to ghrelin. As a result, development of this concept was halted (25), although it is unclear if the antibodies in this study were generated to the essential N-terminal portion of ghrelin, so that the basis of the ineffectiveness of the vaccine remains in question.
Somatostatin in the Suppression of Ghrelin
Somatostatin and its analog octreotide, known to suppress circulating ghrelin levels (26), have failed to reduce appetite in short-term studies of Prader-Willi syndrome (27, 28). The hyperghrelinemia of Prader-Willi syndrome may thus be incidental to appetite and hyperphagia, or somatostatin administration may not adequately address the complexity of the symptomatology. We have documented concomitant somatostatin-mediated suppression of PYY3-36, which may act to neutralize any benefits from ghrelin suppression (27). Studies are in progress to assess the long-term effects of octreotide treatment (29, 30).
PP-fold Hormones
The PP-fold peptide family encompasses NPY, peptide YY (PYY), and pancreatic polypeptide (PP). As noted above, NPY is a key orexigenic neurotransmitter in the hypothalamus; in contrast, PYY and PP act as peripheral signals (Figure 3⇑). These peptides contain a common structural motif, the PP-fold, which consists of a type 2 proline helix and an alpha helix connected by a beta turn (31). These peptides bind to the Y family of receptors, Y1, Y2, Y4, and Y5, all of which are expressed in the hypothalamus as well as in the periphery (32).
Pancreatic Polypeptide
PP is secreted by pancreatic islet cells as well as by neuroendocrine cells in the distal gut. PP is released in proportion to calorie intake, consistent with a role in signaling satiety (33). It has highest affinity for the Y4 receptor, but also binds to Y1 and Y5 (32). There are conflicting data with respect to the circulating levels of PP in obesity. Some groups have found reduced levels, with blunted PP secretion in response to food intake (34, 35) or secretin challenge (36). Other groups have found no significant differences in PP levels between obese and non-obese subjects (37, 38). Moreover, despite weight loss induced by gastric bypass surgery, significant increases in PP levels were not seen (38). These discrepant observations may reflect the confounding factors of impaired glucose tolerance and diabetes, which increases circulating PP levels (36).
PP was originally shown to reduce food intake in mice (39). Repeated intraperitoneal administration of PP in ob/ob mice reduces weight gain and ameliorates insulin resistance and hyperlipidemia (40). Overexpression of PP in the pancreatic islets in transgenic mice causes reductions in food intake and fat mass (41). The exact site of action is not completely known, but PP is thought to signal via the vagus nerve, as vagotomy abolishes the effect of PP in mice (40, 42, 43). In addition to its action on food intake, PP also stimulates sympathetic activity and oxygen consumption, perhaps causing an increased energy expenditure that would augment weight loss (40); however, such an effect is not seen in the transgenic model of PP overexpression (41).
Intravenous infusion of PP, at a dose of ten pmol/kg/min, can reduce food intake in humans. Although PP is known to slow gastric emptying, this dose does not elicit a detectable effect on gastric emptying in infusions into volunteers (44). A PP analog would therefore be predicted to reduce food intake, and two analogs, obinepitide (targeting Y2 and Y4) and TM30339 (targeting Y4 alone) are under development (7TM Pharma). A recent phase I/II double-blind placebo study found that obinepitide inhibits food intake in obese subjects, and a phase II study is underway (45).
PYY3-36
PYY is found in the small and large intestine, the highest levels being present in the colon and rectum. It is released postprandially from the L cells of the gut, where it is co-stored with glucagon-like peptide (GLP)-1 (see below) (46). The release of PYY is proportional to calorie intake, but ingested protein is able to stimulate release more effectively than fat or carbohydrate (47).
The N-terminal truncation of PYY by dipeptidyl peptidase IV (DPP-IV) (48, 49) results in the major circulating species, PYY1-36. Although the full-length form binds to all Y receptor subtypes, PYY3-36 binds to the Y2 and Y5 more selectively (32). The appetite-suppressive effects of PYY are mediated at presynaptic Y2 receptors in the arcuate nucleus, as the anorexigenic effects of PYY are lost in Y2-null mice (50). There is debate surrounding the principal site of action of PYY in the CNS. Batterham and co-workers propose the hypothalamus as the main target of circulating PYY (50). Abbott and colleagues, however, report that vagotomy attenuates the ability of systemically administered PYY to induce c-Fos expression in the hypothalamus, suggesting that the vagus nerve/brainstem complex may predominate as the site of action of PYY (51).
The peripheral infusion of PYY3-36 reduces food intake in rats and humans (50). Although there was some initial concern with regard to the reproducibility of these effects in rodents, the controversy likely stems from incomplete acclimatization of animals in experimental procedures; a stress response will mask the anorectic effect of PYY (52, 53). Moreover, Pyy-null mice, which are hyperphagic and obese, lose body weight (twenty percent) in response to exogenous PYY3-36 (54). In head-to-head comparisons, PYY3-36 is able to reduce food intake and increase thermogenesis (whereas the effect of PYY1-36 on appetite is less clear). DPP-IV cleavage of PYY1-36, therefore, appears important in energy homeostasis (55).
Although some studies have demonstrated lower circulating levels of PYY in obese individuals compared to their lean counterparts (56, 57), this difference is not universally reproduced (58). Obese subjects remain sensitive to the effects of PYY3-36 infusion and respond with reductions in calorie intake comparable to those elicited in lean subjects (56). In our original studies we infused PYY3-36 at physiological levels and found no ill effects, including nausea, although both our group and others have found that infusing higher levels of PYY can cause nausea (59). However, we have found the nausea is usually transient and that the anorectic effect persists even when nausea levels have returned to baseline, suggesting that the anorectic effect is independent of the nausea (Le Roux, unpublished data).
PYY3-36 is therefore an attractive target for obesity treatment, notwithstanding the narrow therapeutic window between appetite suppression and nausea. Amylin Pharmaceuticals has a phase I study of PYY3-36 underway (60), and Nastech, under license from Thiakis, is developing intranasal PYY3-36 (61). Nastech’s dose-ranging study has shown statistically significant effects of nasal PYY3-36 on caloric intake (62). However, a double-blind placebo study by Merck, using Nastech’s formulation, has examined the ability of preprandial nasal PYY3-36 to reduce weight in combination with exercise and a hypocaloric diet over twelve weeks. Patients taking 200 or 600 μg, administered twenty minutes prior to meals, showed no significant decrease in weight despite achieving circulating target concentrations of PYY3-36; significant side effects, principally nausea and vomiting, were observed (63).
Proglucagon Derivatives
Proglucagon is a 160-residue prohormone that undergoes differential, tissue-specific cleavage by prohormone convertases 1 and 2. The major proglucagon cleavage product in the pancreas is glucagon, whereas GLP-1, GLP-2, and oxyntomodulin (OXM) are expressed in the brainstem and intestine (64). OXM and GLP-1, along with PYY, are released from intestinal L-cells in response to ingested nutrients and are thought to act as short-term satiety signals, possibly participating in long-term body weight regulation as well.
GLP-1 and GLP-1 Analogs
The major circulating form of GLP-1 is GLP-1(7-36) amide, although GLP-1(7-37) is biologically equipotent (65, 66). Together with glucose-dependent insulinotropic polypeptide (GIP), GLP-1 stimulates the expression and secretion of insulin following oral glucose administration (i.e., the incretin effect). It also acts as an anti-apoptotic factor for pancreatic beta cells (67, 68). Native GLP-1(7-36) amide is subject to rapid breakdown and inactivation in the circulation by the actions of DPP-IV (69).
The postprandial release of GLP-1 occurs rapidly, with carbohydrate and fat each acting as stimulants to secretion (70). Mono-unsaturated fatty acids are particularly powerful in stimulating GLP-1 secretion, which may account for the beneficial effects of diets containing these fatty acids on glycemic control in patients with diabetes (71). A complex neuro-humoral system regulates secretion of GLP-1, involving vagus nerve afferents and efferents, the enteric nervous system, and GIP, as well as direct nutrient contact with L-cells in the distal gut (72). The potent incretin effect of GLP-1 has received much attention from both a physiological and a therapeutic viewpoint (73). More recently, the effects of GLP-1 on energy homeostasis have also proven to be of great interest. GLP-1 inhibits food intake in rats (74). In humans, GLP-1 has been shown to slow gastric emptying and to lengthen the period of postprandial satiety (75, 76).
GLP-1 Analogs for Treatment of Diabetes and Obesity
Because GLP-1 is broken down quickly in the circulation by DPP-IV, Amylin Pharmaceuticals have developed a more stable analog for pharmacotherapy. Isolated from the venom of the Gila monster lizard, exendin-4 (Byetta/exenatide) is a DPP-IV–resistant peptide agonist at the GLP-1 receptor; its circulating half-life is eightyfold greater than that of GLP-1 (77). Even so, it is necessary to administer exenatide subcutaneously twice a day. Exenatide is licensed in the US and the EU as an adjunct to metformin and/or sulphonylurea therapy for type 2 diabetes and has been shown to improve glycemic control and reduce body weight (78–81). Notably, obese type 2 diabetes patients on insulin also benefit from the addition of exenatide, with significant weight loss, reduction in insulin requirements, and improvement in glycemic control and other metabolic parameters (82). Nausea is the most significant side effect, affecting twenty-fine to sixty percent of patients but accounting for only a minority of discontinuations (83). Recently, a concern has been raised about the association between exenatide therapy and acute pancreatitis, based on thirty cases detected in post-marketing surveillance (84). A long-acting release (LAR) formulation of exenatide, for weekly subcutaneous injection, is under development (85), in which the drug is adsorbed to polylactide-glycolide microspheres. In a fifteen-week trial of diabetic patients who had responded poorly to metformin or diet/exercise, high-dose exenatide LAR was effective in improving glycemia as well as in reducing body weight. Interestingly, lower doses, which have a significant effect on carbohydrate tolerance, show no effect on weight (85). Exenatide LAR is currently in phase II trials.
Another GLP-1 analog, liraglutide, has been developed by Novo Nordisk and is nearing approval for marketing in 2008. This peptide-based analog, partially resistant to DPP-IV, bears a covalent sixteen-carbon fatty acid that enables the drug to bind to circulating albumin, in a manner similar to Novo Nordisk’s insulin levemir. Liraglutide has an approximate half-life of eleven to fifteen hours after subcutaneous injection, allowing for once-daily injection (86). When administered with metformin in type 2 diabetics, liraglutide improves glycemic control over five weeks (87). Phase III studies confirm that liraglutide, administered with either metformin or sulphonylurea, is better than rosiglitazone at improving glycemic control (88). In a phase II study of obese patients without diabetes, liraglutide was significantly more effective than orlistat or placebo in promoting weight loss. It must be noted that thirty percent of the patients in this study, which lasted over twenty weeks, had prediabetes (i.e., impaired glucose tolerance or impaired fasting glucose). Liraglutide improved systolic blood pressure, and compared to either placebo or orlistat, was far more effective in resolving prediabetes. The most common side effect was nausea (89).
The fusion of recombinant GLP-1 to albumin results in a protein (albiglutide) that has a circulating half-life extended relative to that of GLPL-1. Developed by Human Genome Sciences and GlaxoSmithKline and currently in phase II evaluation, albiglutide has been shown to activate the GLP-1 receptor and to reduce food intake in mice, although to a lesser extent than does exenatide (90). A similar drug developed by Conjuchem, CJC-1131, uses a DPP-IV–resistant GLP-1 derivative that contains a reactive chemical linker. Once parenterally administered, this linker reacts with albumin to form a stabilized conjugate, which has an incretin effect in mice but does not reduce weight in diabetic db/db mice (91). Weekly administration of an exendin-4–albumin conjugate, CJC-1134-PC, is also under phase II investigation (92). Lastly, Ipsen and Roche are developing a GLP-1 analog (BIM 51077) that is in phase II development (93), but few data concerning its effectiveness are available.
DPP-IV Inhibitors
DPP-IV inhibitors increase circulating levels of GLP-1 by preventing its breakdown. One orally active DPP-IV inhibitor, sitagliptin, is currently marketed as Januvia (Merck). A second inhibitor, vildagliptin (Glavus), has been submitted by Novartis for approval to the FDA. Sitagliptin inhibits DPP-IV by at least eighty percent over twenty-four hours and increases circulating active GLP-1 levels (94). In an eighteen-week trial of patients with diabetes not adequately controlled by diet and exercise, sitagliptin seginificantly reduced glycosylated hemoglobin (HbA1c; 0.48–0.60%), the only notable side effect being nausea (one to two percent) (95). A double-blind controlled trial of vildagliptin over twelve weeks in diabetic patients treated with metformin demonstrated that this DPP-IV inhibitor also was able to reduce HbA1c (by 0.7% versus placebo) (96). Despite modest benefits on glycemic control in type 2 diabetics, neither trial demonstrated weight loss. It must be noted that inhibition of DPP-IV may have multiple, perhaps conflicting, effects on metabolism, many of which warrant investigation (97).
Oxyntomodulin
OXM is another product of preproglucagon processing and is released into the circulation after a meal (98). Like GLP-1, it reduces food intake and body weight gain in rats (99, 100). Similarly, peripheral adminstration of OXM in humans reduces energy intake and hunger, without causing nausea, as judged by visual analog scales (101). The anorectic effects of OXM are blocked by the GLP-1 receptor antagonist exendin 9–39 (100) and do not manifest in GLP-1 receptor knockout mice (102). The peripheral administration of OXM, as judged by c-Fos expression in hypothalamic nuclei, also reflects a pattern of neuronal activation elicited by GLP-1 (102); however, OXM appears to activate CNS pathways distinct from those engaged by GLP-1. Manganese-enhanced MRI reveals different patterns of neuronal activation in the hypothalamus in response to GLP-1 and OXM (103). Moreover, the intra-arcuate injection of exendin 9–39 blocks the anorexigenic effect of intra-arcuate OXM but does not block the effect of GLP-1 (100). Small double-blinded studies of OXM have been conducted in obese subjects. Subcutaneous administration three times daily over four weeks reduces energy intake and increases weight loss (104); activity-based energy expenditure is increased by about twenty-six percent, and total energy expenditure by about nine percent (105). OXM thus remains an attractive area for pharmacotherapeutic investigation. As with GLP-1, OXM is degraded by DPP-IV (106, 107) and is therefore rapidly cleared from circulation after subcutaneous injection. An OXM analog, resistant to rapid degradation by DPP-IV, is currently under development by Thiakis Limited (108).
Current Anti-Obesity Therapies
The most effective treatment available for achieving and sustaining weight loss is bariatric surgery, but both cost and mortality preclude this treatment for the majority of the obese population. Thus, the search for a safe and user-friendly pharmacological responses to the obesity epidemic remains urgent. The solution is likely to come from manipulating the body’s own regulatory pathways for energy homeostasis. The therapeutic exploitation of gut hormones is one avenue under exploration, but the disadvantages of these agents include short duration of action, the necessity for parenteral administration, narrow therapeutic windows, and nausea.
Cholecystokinin
Cholecystokinin (CCK) is in reality a group of peptides derived from preproCCK by prohormone convertase and carboxypeptidase E. CCK peptides are sulphated and terminally amidated, and they share a common active C-terminal heptapeptide [Tyr(SO3H)-Met-Gly-Trp-Met-Asp-Phe-NH2] (109). In response to food intake (110), secretion of CCK [by I-cells, principally in the duodenum and jejunum (111)] classically results in contraction of the gallbladder, exosecretion by the pancreas, and relaxation of the sphincter of Oddi. However, CCK is also to be found in the pituitary (in corticotrophs), the enteric nervous system, and cortical areas of the brain. In these latter areas, CCK functions as a neurotransmitter. Two 7-transmembrane domain CCK receptors, CCK1 (CCK-A) and CCK2 (CCK-B), are found in the gut and in the brain and pituitary (109).
In 1973, CCK became the first gut hormone to be recognized for eliciting satiety in rats (112). This effect is abolished by vagotomy, indicating that the effect is mediated via CCK receptors on the vagus nerve (113). Infusion of the C-terminal octapeptide of CCK reduces food intake in obese men (114), prompting GlaxoSmithKline to develop a selective CCK1 receptor agonist (GI181771X). Unfortunately, monotherapy with GI181771X elicited no significant changes in the weight or metabolic parameters in the clinic (115), a finding that may stem from the fact that CCK’s effects are subject to tachyphylaxis (116).
Amylin
Also known as islet cell amyloid peptide, this thirty-seven–residue peptide is normally co-secreted with insulin by beta cells (117). Interestingly, amylin does not have its own unique receptor, but instead binds to a receptor complex consisting of a calcitonin receptor plus a protein modulator; six distinct amylin receptor activities have been characterized (118). Amylin retards gastric emptying (119) and inhibits L-arginine–stimulated glucagon secretion (120). Amylin is secreted in proportion to food intake (121) and acts as a satiety factor. Injection of amylin inhibits food intake in rats (121–123), apparently by acting directly on the area postrema to modulate CCK signaling (124–125). A synthetic analogue of amylin, (pramlintide; Symlin–Amylin Pharmaceuticals), is licensed for therapy of type 1 and type 2 diabetes in the United States but not in Europe. As an adjunct to insulin therapy, pramlintide has modest effects on body weight, eliciting the loss of 0.5–1.0 kg over one year in patients with type 1 diabetes (126) and 1.4 kg in patients with type 2 diabetes (127).
Although it is possible to achieve modest weight loss by targeting GLP-1, PYY3-36, OXM, and amylin, significant improvements in morbidity and mortality will be necessary to achieve meaningful weight loss for obese populations. The experience of bariatric surgery gives us some important clues as how to achieve this goal. Post surgery, significant weight loss is associated with elevations in GLP-1, OXM, and PYY3-36, whereas ghrelin levels are not altered (128). These observations likely underlie the reduction in hunger reported by patients long before substantial weight loss occurs. Optimal therapy may thus require a synergistic combination of hormonal agents, each administered at the appropriate dose. Research to establish correct synergistic combinations has not reached fruition but is ongoing. For example, the co-administration of PYY3-36 and GLP-1 additively inhibit food intake in humans (129). Amylin and PYY3-36 act synergistically to reduce food intake in rats but have additive effects with respect to weight loss (130). The development of peptide analogs with enhanced resistance to degradation and clearance, along with new small-molecule agonists and antagonists, promises to expand our options in anti-obesity therapies. New drug delivery technologies, such as intra-nasal sprays, may soon mitigate the inconvenience of injections. Finally, by fine-tuning the pharmacokinetics and delivery of these drugs, we may yet be able to minimize side effects while reducing energy intake, increasing energy output, and sustaining losses in body weight.
Current Pharmacotherapy of Obesity
Our current understanding of the points at which anti-obesity drugs might be employed to modify energy homeostasis is outlined in Figure 2⇑. Appetite, which determines the amount of energy ingested, is regulated by two central nervous system networks. The first is a “homeostatic” system that stabilizes body fat mass by monitoring meal-related signals from the gastrointestinal tract as well as adiposity signals proportional to the total amount of triglyceride stored in adipocytes. This system is the target of sibutramine, one of three drugs approved for the long-term treatment of obesity. The “hedonic” system is the second central network; it attaches reward value to a variety of behaviors (e.g., food ingestion). The hedonic system is the target of rimonabant, a drug currently approved for treating obesity in Europe. Normally, nearly all of the energy ingested in food is absorbed by the gastrointestinal tract. Orlistat, the third approved anti-obesity drug, interferes with the processing of triglyceride in the intestine and thereby reduces gastrointestinal absorption of energy. No pharmacological agents are available that directly block the deposition of absorbed energy as triglyceride in adipocytes or that increase either the voluntary or involuntary components of energy expenditure.
Sibutramine blocks the presynaptic reuptake of both serotonin and norepinephrine in the central and peripheral nervous receptors, systems, thereby activating POMC neurons, via 5-HT2C in the arcuate nucleus of the hypothalamus (131). POMC neurons play a key role in energy homeostasis by delivering anorectic signals to the hypothalamic paraventricular nucleus and other critical brain areas (Figure 3⇑). Thus, sibutramine causes a decrease in energy ingestion that results in modest weight loss, along with a sympathetic response that results in modest increases in heart rate and blood pressure. A recent meta-analysis of ten double-blind randomized placebo-controlled trials (n = 2,623) found that sibutramine reduces body weight by approximately four percent over the course of twelve to eighteen months (132). Sibutramine also increases (by approximately eighteen percent, relative to placebo) the percentage of trial subjects who achieve a ten-percent reduction in body weight (132).
Orlistat inhibits the gastrointestinal lipases, predominantly pancreatic lipase, that hydrolyze triglycerides into free fatty acids in the intestine. Interference with this key reaction can result in failure to absorb up to thirty percent of energy ingested as fat, which is instead passed out in the feces. Weight loss ensues both from the reduction in energy transiting the gastrointestinal mucosa and from the tendency of successful users to avoid fat ingestion due to the unpleasant consequences of fat malabsorption. In sixteen double-blind randomized placebo-controlled trials involving 10,631 participants, orlistat elicited the loss of approximately three percent body weight relative to placebo over the course of one to four years. Orlistat also increased (by about twelve percent, relative to placebo) the percentage trial subjects who achieve a ten-percent reduction in body weight (132).
Rimonabant: Endocannabinoid System Blockade
Rimonabant is the first of a new class of drugs that block the overactivity of the endocannabinoid system in overweight and obese patients. Having both a central and a peripheral action, these drugs help not only to control weight, but also to improve lipids and insulin sensitivity in patients with the metabolic syndrome. Other endocannabinoid receptor blockers and/or reverse agonists are in clinical trials, and their efficacy and safety will soon be known. As a class, these agents may play an important role in the treatment of obesity and its metabolic complications.
The trials of sibutramine and orlistat summarized above probably overestimate the effectiveness of these agents in real clinical practice for several reasons. First, subjects had been selected for responsiveness to the hypocaloric diet used as adjunctive therapy, so that only the most motivated individuals were recruited as trial candidates. In addition, study subjects generally receive a much higher level of ongoing support and supervision than do patients in the practicing clinic, and they are therefore more likely to comply with treatment. Finally, trials may not correct for the effect of subject dropout rates [about thirty to forty percent (132)]. In a recent survey of nearly 17,000 orlistat users and 3,500 sibutramine users in Canada, the rates of ongoing use of either drug were less than ten percent at one year and two percent at two years (133). These very high attrition rates reflect both the failure of the drugs to produce the desired degree of weight loss and the side effect profiles of the drugs.
The clear message, from both the historical drug failures discussed previously and experiences with currently available drugs, is that drug side effect and safety profiles must receive exceedingly close scrutiny. Such vigilance is particularly important in light of the vast number of candidates for obesity pharmacotherapy. Some potential toxicities of current and future anti-obesity drugs can be predicted from the scheme outlined in Figure 2⇑. Agents that affect homeostatic mechanisms of appetite reduction might also be expected to produce nausea, the most extreme manifestation of satiety. Agents that reduce the hedonic value of eating may reduce the hedonic value of many other experiences as well, resulting in depression. Agents that cause nutrient malabsorbtion risk gastrointestinal side effects as well as potential vitamin and mineral deficiencies. Agents that block adipogenesis or the uptake of fat by existing adipocytes may divert fat to liver and muscle, where fat storage can produce insulin resistance and hyperlipidemia. Finally, agents that increase energy expenditure may cause hyperthermia and increase tissue damage due to reactive oxygen species. In addition, it is difficult to justify drugs that would simply lead to wastage of the ingested food energy that our planet is already so hard-pressed to supply.
Experience with currently available drugs indicates that weight loss induced by any single agent rarely exceeds ten percent of starting weight, and after six months the statistic drops to five percent, even among people who carry 100–150 percent excess body fat. This “ceiling effect” in monotherapy likely reflects the high degree of redundancy among regulatory pathways for energy intake and expenditure. Inhibition of one pathway leads to weight loss only until compensatory changes occur in other pathways. Successful pharmacotherapy of obesity will probably require drugs to target key steps in multiple energy homeostatic pathways, just as cancer chemotherapy requires multiple agents to inhibit tumor cell growth. Thus, pharmacotherapy as a sole approach to the obesity problem may prove exceedingly expensive, exposing patients to a large number of drug side effects.
The Endocannabinoid System as an Emerging Target
The endocannabinoid system consists of receptors, their endogenous ligands, and the enzymes that synthesize and degrade these ligands. Overactivation of the endocannabinoid system is associated with increased food intake, obesity, and dyslipidemia (134, 135). Cannabinoid 1 (CB1) receptors, which are G protein–coupled receptors, are present in the central nervous system and peripheral sites, including adipose tissue, liver, gastrointestinal tract, pancreas, muscle, and the cardiovascular system. Upon agonist binding, CB1 receptors inhibit neurotransmitter release in central and peripheral neurons, including effects on neurons of the mesolimbic system that participate in reward behaviors. Peripheral effects of CB1 receptor activation are widespread, including increased hepatic lipogenesis, increased fatty acid synthesis in adipose tissue, and downregulation of adiponectin production.
Rimonabant is a selective CB1 antagonist/reverse agonist (136, 137), developed for the management of body weight and multiple cardiovascular risk factors. Four phase III studies, comprising the Rimonabant in Overweight/Obesity (RIO) program, have assessed the efficacy and safety of rimonabant. The RIO-Europe (n = 1507) and RIO-North America (n = 3040) trials were two-year, multicenter, randomized, double-blind, placebo-controlled parallel-group studies, comparing placebo with rimonabant in overweight or obese patients on a low-calorie diet. The primary endpoint was weight loss, and secondary endpoints included waist circumference, lipid levels, fasting glucose and insulin, and metabolic syndrome prevalence.
Patients treated daily for one year with 20 mg rimonabant in the RIO-Europe study lost an average of about 6.6 kg (compared to 1.8 kg for those on placebo). Over one-quarter of patients on this regimen of rimonabant (compared to less than eight percent of patients on placebo) lost more than ten percent of their initial body weight and had an average decrease in waist circumference of 8.5 cm (versus 2.4 cm for those on placebo). In addition to weight loss, a statistically significant improvement in metabolic risk factors was observed. Side effects were generally mild and transient, the most common of which were nausea, diarrhea, and dizziness (137). Only in a very small number of cases did these side effects lead to discontinuation of drug use (138).
The RIO-North America trial similarly established efficacy, with significantly lowered weight, reduced abdominal fat, diminished cardiovascular risk factors, and decreased metabolic disorders among patients treated with the daily 20-mg dose (138). Weight loss data are shown in Figure 4⇓; effects on waist circumference (data not shown here) were similar. At one year, weight loss was about 6.3 kg for the average patient on 20 mg rimonabant per day (compared to 1.6 kg among the placebo group), and waist circumference was reduced by 6.1 cm (vs 2.5 cm on placebo). Over forty-eight percent of the drug-treated patients (vs twenty percent on placebo) lost more than five percent of initial body weight; 25.2% of patients on drug (vs 8.5% on placebo) lost over ten percent body weight. Patients who remained on rimonabant for a second year maintained their weight loss, whereas those who were re-assigned to placebo regained their weight (Figure 4⇓). Metabolic measures were also significantly improved by rimonabant treatment throughout the two-year period. HDL-cholesterol increased by 24.5% in the rimonabant (20 mg daily) group, compared to 13.8% in the placebo group; triglycerides were reduced by 9.9% among treated subjects (vs 1.6% on placebo). Rimonabant significantly improved insulin sensitivity and resolved the metabolic syndrome of one-third of those patients so diagnosed at study outset.
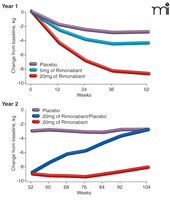
Effect of rimonabant on body weight (RIO-North America trial). In the second year of the trial, subjects who had been on the 20-mg daily dose were randomly re-assigned to either continue treatment or switch to placebo. The observed weight loss was also reflected in reduction of waist circumference (data not shown). [Data reproduced from (7). See text for details.]
The Rio-Lipids study was a one-year, randomized, double-blind clinical trial in overweight or obese patients with untreated dyslipidemia and without diabetes (139). Subjects taking 20 mg of the drug daily (on a low-calorie diet) lost an average of 6.7 kg and 5.4 cm in waist circumference; showed a ten-percent increase in HDL-cholesterol; experienced a thirteen-percent decrease in triglycerides; showed significant reductions levels of C-reactive protein and small, dense LDL-cholesterol particles; and manifested an increase of 57.7% in plasma adiponectin. The Rio-Diabetes trial, also conducted for one year, assessed overweight patients with type 2 diabetes mellitus (HbA1c approximately 6.5 to 10%) taking metformin or sulfonylureas(139, 140). Patients receiving 20 mg drug daily underwent a weight loss of 3.9 kg and a reduction of waist circumference of 3.3 cm. The change of HgA1c was 7.3% to 6.7% [as opposed to 7.2% to 7.3% on placebo (p<0.001)]; among patients with HbA1c >8%, rimonabant elicited a reduction of 1.1%. HDL-cholesterol increased 8.4%; triglycerides dropped 16.4%; blood pressure dropped 2.3 mm Hg; and C-reactive protein decreased 25% (140).
Fatty Acid Metabolism in Anti-Obesity Research
Modulation of fatty acid metabolism as a treatment strategy for obesity is still in its infancy, although a number of substantial observations have been made. The enzymes of fatty acid synthesis are highly expressed in hypothalamic nuclei critical to modulating feeding behavior. FAS inhibition has been shown to modulate hypothalamic neuropeptides, reducing food intake and leading to weight loss in rodents. Recently, the pharmacological stimulation of fatty acid oxidation has been shown to reduce food intake, an observation consistent with the longstanding finding that the inhibition of fatty acid oxidation stimulates food intake. The target-rich pathways of fatty acid metabolism will likely provide a number of strategies for alteration of both energy metabolism and food intake.
Inhibition of Fatty Acid Synthesis
Obesity can be viewed as an overall disorder of energy balance, resulting from dysreglulation of both appetite and metabolism (141–143). The regulation of energy balance, essential to all organisms, is managed by many redundant pathways. This redundancy, although crucial to species survival, nevertheless signifies that pathophysiological perturbations of energy balance cannot be remedied by altering any single signaling system. The role of fatty acid metabolism in the regulation of energy balance in the brain and periphery has become an area of intense interest. Studies from Ronnett and colleagues have focused both on inhibiting fatty acid synthesis, with fatty acid synthase (FAS) as the target, and on enhancing fatty acid oxidation; the latter focus centers on carnitine palmitoyltransferase-1 (CPT-1), the rate-limiting enzyme for the entry of fatty acids into the mitochondria for oxidation, and glycerol-3-phosphoacyltransferase (GPAT), the initial enzyme in acylglyceride synthesis. Inhibition of FAS or stimulation of CPT-1, using distinct pharmacological agents, reduces food intake and incurs profound reversible weight loss. Recently, the modulation of fatty acid metabolism in vivo has been unambiguously linked to key regulatory mechanisms in energy metabolism and feeding behavior, leading to reduced animal weight and adiposity (144–149).
Fatty Acid Synthase Inhibition
FAS promotes the de novo synthesis of fatty acid in the cytosol (Figure 5⇓), catalyzing the NADPH-dependent reductive synthesis of palmitate from acetyl-CoA and malonyl-CoA (150). Malonyl-CoA provides the carbon for successive rounds of condensation during fatty acid elongation. In mammals, FAS is the product of a single gene that produces a 250-kD protein that functions as a dimer, simultaneously synthesizing two molecules of palmitate (151). FAS provides the means for converting excess dietary carbohydrate into fat, thereby preserving ingested calories that are not immediately needed for metabolic processes. During periods of energy excess, fatty acids are esterfied to glycerol and stored in the liver and adipose tissue as triglycerides. The implication of fatty acid metabolism in the modulation of food intake originated in studies of human cancer, which is now understood to be lipogenic; FAS is highly expressed in many common cancers (152). Unlike normal lipogenic tissues, such as liver and adipose tissue, human cancers synthesize fatty acids but do not store fat as triglyceride. Instead, fatty acid synthesis functions in cancer cells to maintain redox and energy balance (153). The first synthetic FAS inhibitor, C75, designed to probe tumor cell apoptosis, manifested weight loss as its “dose-limiting toxicity” (154, 155).
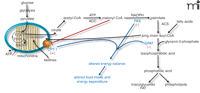
Enzyme targets in fatty acid metabolism. Inhibition of fatty acid synthesis, by inhibiting fatty acid synthase (FAS), and enhancing fatty acid oxidation, by targeting carnitine palmitoyltransferase-1 (CPT-1) or glycerol-3-phosphoacyl-transferase (GPAT), can effect dramatic changes in hypothalamic energy balance. Significant weight loss through modulation of these enzymes reflects reduced food intake and increased energy expenditure. (See text for details. ACC, acetyl CoA carboxylase; ACS, acyl CoA synthetase; TCA, tricarboxylic acid.)
The effect of FAS inhibition on feeding behavior reflects an influence on neural pathways responsible for appetite control, specifically in the hypothalamus (156–158). Although originally localized to non-neuronal cells in the brain (159), subsequent studies demonstrated that FAS was actually expressed in neurons (145). FAS is highly expressed in human hypothalamus, with particularly high levels in the arcuate nucleus and other hypothalamic nuclei associated with feeding. Importantly, brain FAS is regulated quite differently from peripheral FAS. In the brain, FAS message and protein levels are unaffected after twenty-four hours of starvation, while FAS protein and message become undetectable in the liver(145). FAS inhibition leads to profound changes in hypothalamic neuropeptides, reducing the expression of the orexigenic neuropeptides NPY and AgRP while enhancing the expression of anorexigenic neuropeptides such as CART (see Figure 2⇑) (147, 160–162). These effects on hypothalamic neuropeptides are manifested, in lean and diet-induced obese mice, as decreased food intake and body weight.
Fatty Acid Oxidation Stimulation
Intriguingly, the pharmacological inhibition of fatty acid oxidation, at multiple enzyme targets, does not inhibit, but rather stimulates, food intake (163–166). One of these targets, CPT-1, is the rate-limiting enzyme for mitochondrial fatty acid oxidation (167). CPT-1 governs the transport of fatty acids into the mitochondrial matrix, where they are irrevocably oxidized. Regulation of CPT-1 is thus essential for precluding the futile cycling of newly synthesized fatty acids into the mitochondrion; CPT-1 activity is competitively inhibited by malonyl-CoA, the key substrate of FAS (Figure 5⇑). More recently, studies with C75 and new pharmacological agents that selectively and directly stimulate CPT-1 activity, have shown that the facilitation of fatty acid oxidation effectively reduces food intake (148, 149, 168). This decline in food consumption occurs whether the agents are administered systemically or intracere-broventricularly (168–170). The stimulation of fatty acid oxidation appears, like FAS inhibition, to affect food intake via central neural pathways that may involve hypothalamic signaling.
The pharmacological inhibition of mammalian GPAT (Figure 5⇑) has also been addressed recently by Ronnett and colleagues. GPAT, existing in at least three isoforms, esterifies fatty acids to glycerol-3-phosphate (171). Preliminary studies show that GPAT inhibition in vivo leads to both increased fatty acid oxidation and reduced food intake. These findings are reminiscent of CPT-1 stimulation. These data suggest that regardless of the biochemical pathway involved, if fatty acid oxidation is enhanced, increased energy expenditure and reduced food intake will ensue.
The Future of Obesity Pharmacotherapy
The complex molecular pathways involved in body weight regulation represent a large number of potential targets for new drugs to treat overweight and obesity. But if the global obesity epidemic reflects a failure to create a healthy environment, rather than a failure of our physiology per se, what role should drugs play in treating a condition that already affects 1.1 billion people worldwide (171) and continues to increase in prevalence? In fact, it is the robust operation of homeostatic mechanisms defending body fat stores that makes it so difficult to treat established obesity. Environmental factors promoting obesity include the ready availability and heavy marketing of inexpensive energy-dense food, an infrastructure that deters the incorporation of physical activity into one’s daily routine, and a proliferation of sedentary leisure time activities. Presumably, obesity could be prevented if these adverse environmental trends were reversed, circumventing the need for pharmacological approaches to the problem.
Despite the need for effective public health measures, the development of more effective anti-obesity drugs is justified for at least two reasons. First, physiological defects will account for a subset of individuals with refractory obesity, best addressed through proper molecular intervention. For example, roughly five percent of individuals with severe childhood-onset obesity carry mutations that interfere with the function of the melanocor-tin-4 receptor (MC4R) (172). These individuals might be treated effectively by an MC4R super-agonist, or by a drug that activates signaling pathways distal to MC4R. As our knowledge of energy homeostasis grows, it is likely that additional subgroups of obese individuals will be identified as candidates for targeted pharmacological interventions.
The second justification for new anti-obesity drugs is the likelihood that lifestyle modifications, even if widely implemented, will be relatively ineffective in treating obese individuals already born into the current metabolically toxic environment. There is increasing evidence that environmental factors such as maternal overnutrition and hyperinsulinemia, and excessive caloric intake during the first months of life, can alter the connectivity of critical hypothalamic circuits in the fetus and neonate (173). These alterations may program energy homeostatic mechanisms to preserve a higher body fat mass throughout life. If this is true, then it may take several generations of aggressive public health intervention to reset the population to the average body fat mass that typified previous generations. During the present, hopefully limited, period beset by obesity, intensive drug treatment may be necessary for many individuals to control elevated body weight and associated cardiovascular risk factors. As discussed earlier, this control will probably require multiple expensive drugs, targeting redundant energy homeostasis pathways. It will be incumbent upon the health care system to make these drugs available to members of all social classes, including the economically disadvantaged minorities that bear a disproportionate share of the burden of obesity.
- © American Society for Pharmacology and Experimental Theraputics 2008
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
-
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵

D. Scott Weigle, MD, is Professor of Medicine at Harborview Medical Center in Seattle. His studies of experimental and human obesity concern the mechanisms and signaling factors that influence appetite and fuel metabolism.

Tricia Tan, MD, qualified in St Andrew’s and Manchester University, and trained in Endocrinology, Diabetes and Internal Medicine in London. Her research interests lie in the regulation of appetite by ghrelin and pancreatic polypeptide. She is currently Clinical Research Fellow and Honorary Consultant at Imperial College Healthcare NHS Trust.

Gabriele Ronnett, MD, PhD, is Professor of Neuroscience at the Johns Hopkins University School of Medicine. Her research has exploited the olfactory system to advance our understanding of neuronal development and of developmental diseases. She also studies the role of brain signaling pathways in regulating energy balance and food intake.

F. Xavier Pi-Sunyer, MD, is Professor of Medicine at Columbia University College of Physicians and Surgeons and Professor of Applied Physiology at Columbia Teachers College. He is Chief of Endocrinology, Diabetes, and Nutrition at Luke’s-Roosevelt Hospital Center and Senior Attending Physician, and is also Director of the New York Obesity Research Center. His research has greatly advanced clinical approaches to carbohydrate and lipid metabolism, obesity, and diabetes.

Ismail Laher, PhD, organized the Experimental Biology 2008 symposium on which this article is based. He is Professor of Pharmacology and Therapeutics at the University of British Columbia. Address correspondence to IL. E-mail ilaher{at}interchange.ubc.ca.

Francis P. Kuhajda, MD, is Professor of Pathology and Oncology at the Johns Hopkins University School of Medicine. His research focuses on fatty acid metabolism and its potential clinical manipulation in cancer and obesity.

Stephen R. Bloom, MD, is Professor of Medicine at the Imperial College London and directs the Division of Investigative Science. He is also the clinical director for pathology and therapy services at Hammersmith Hospital in London. His expertise includes the neuropeptide-mediated regulation of food intake the regulatory pathways that underlie experimental and clinical approaches to obesity.

