|
 
 |
|
ORIGINAL ARTICLE |
|
|
|
| Year : 2012 | Volume
: 18
| Issue : 2 | Page : 193-197 |
| |
Molecular epidemiology of β-thalassemia in Pakistan: Far reaching implications
Saqib H Ansari, Tahir S Shamsi, Mushtaq Ashraf, Tasneem Farzana, Muneera Bohray, Kousar Perveen, Sajida Erum, Iqra Ansari, Muhammad Nadeem Ahmed, Masood Ahmed, Faizan Raza
Department of Pediatric Hematology & Molecular Medicine, National Institute of Blood Diseases, Karachi, Pakistan
| Date of Web Publication | 8-Sep-2012 |
Correspondence Address:
Saqib H Ansari
Paediatric Haematologist, National Institute of Blood Diseases, ST 2/A, Block 17, Gulshan-e-Iqbal, KDA Scheme 24, Karachi
Pakistan
 Source of Support: None, Conflict of Interest: None  | 1 |
DOI: 10.4103/0971-6866.100762

 Abstract Abstract | | |
Background: β -Thalassaemia, an autosomal recessive hemoglobinopathy, is one of the commonest genetically transmitted disorders throughout the world. Collective measures including carrier identification, genetic counseling and prenatal diagnosis are required for preventing β-thalassemia.
Aim: To achieve this objective, Identification of the spectrum of genetic mutations, especially for various ethnic backgrounds in Pakistan. Therefore, we designed a cross sectional prospective study to identify the frequency of various gene mutations in different ethnic groups of Pakistan.
Materials and Methods: Over a 5-year period, DNA from 648 blood samples {including specimens of chorionic villus sampling (CVS)} were analyzed for the twelve most common β-thalassemia mutations found in the Pakistani population by a Multiplex amplification refractory mutation system (ARMS). Each sample was analyzed for the mutation as well as the normal gene, appropriate with negative and positive controls, and reagent blanks.
Results: Out of 648 samples mutations were identified in 640 (98.75%) samples by multiplex ARMS. 8 common β-thalassemia mutations were identified in 8 different ethnic groups accounting for 93.9% of the β-thalasemia alleles.
Conclusions: Based on the outcome of this study a cost effective proposal is formulated for detection of β-thalassemia mutations.
Keywords: Gene frequency, genetic epidemiology, prenatal diagnosis, thalassaemia prevention
How to cite this article:
Ansari SH, Shamsi TS, Ashraf M, Farzana T, Bohray M, Perveen K, Erum S, Ansari I, Ahmed MN, Ahmed M, Raza F. Molecular epidemiology of β-thalassemia in Pakistan: Far reaching implications. Indian J Hum Genet 2012;18:193-7 |
How to cite this URL:
Ansari SH, Shamsi TS, Ashraf M, Farzana T, Bohray M, Perveen K, Erum S, Ansari I, Ahmed MN, Ahmed M, Raza F. Molecular epidemiology of β-thalassemia in Pakistan: Far reaching implications. Indian J Hum Genet [serial online] 2012 [cited 2016 Jun 1];18:193-7. Available from: http://www.ijhg.com/text.asp?2012/18/2/193/100762 |
| Introduction | |  |
Thalassemia comes from a Greek word "Thalas0" meaning the sea and "emia" for blood. The word came into use as thalassemia was originally described in countries bordering the Mediterranean Sea. It was not recognized as an entity till 1925 when Cooley and Lee described a syndrome occurring early in life associated with splenomegaly and bony deformities. Thalassemias are a heterogeneous group of inherited disorders of hemoglobin synthesis, resulting in life-threatening anemia and requiring regular blood transfusion for survival. [1] It's particularly associated with people of Mediterranean, Indian subcontinent, and Middle East origin. The World Health Organization (WHO) has identified control of hemoglobinopathies, particularly β-thalassemia, in developing world as a priority. [2] An estimated 5000-9000 children with β-thalassemia are born per year, although no documentary registry is available in Pakistan. The estimated carrier rate is 5-7%, with 9.8 million carriers in the total population. [3]
With gradual control of malnutrition and communicable diseases, β-thalassemia major patients who earlier died young are now surviving long enough to seek medical attention. In developing countries like Pakistan, this poses an increasing burden for health-care services as adequate blood transfusions with effective iron chelation and bone marrow transplantation is affordable for just a few; therefore, prevention has been demonstrated to be the way forward. Effectiveness of a 20-year control program in Sardinia is evidenced by reduction of the birth rate of thalassemia major from 1:250 live births to 1:4000. [4] In 1995, 1999 and 2004, 296, 94 and 56 β-thalassemia homozygote, respectively, were born (2.53, 1.07 and 0.82 patients per 1000 births) - thanks to a 10-year program in southern Iran. [5] Population screening, genetic counseling, prenatal diagnosis, and option of terminating affected pregnancies remain the mainstay strategy to devise a control program, and investigating the underlying molecular defects in β-thalassemia is an important prerequisite for such programs.
Currently, 217 causative molecular defects have been described so far in the β-globin gene causing β-thalassemia. [6] About 20 mutations account for 90% of β-globin genes in the world, and it is noted that each ethnic population has its own unique set of most frequent mutations. Previously, there have been few studies investigating the spectrum of β-thalassemia mutations in various regions and ethnic groups of Pakistan. [7],[8] The aim of this study is to identify the frequency of various mutations in Karachi, the largest cosmopolitan city of Pakistan with a population in excess of 10 million, having a significant representation of all the major ethnic groups and to formulate a comprehensive and affordable mutation detection strategy for control of β-thalassemia.
| Materials and Methods | | |
The study was conducted at Department of Pediatric Hematology and Molecular Medicine, National Institute of Blood Diseases (NIBD) in Karachi. The study protocol was approved by the ethics committee at NIBD, and an informed consent was taken from all patients / parents. The studied population included members of the 5 major ethnic groups in Karachi: Punjabi, Pathan, Sindhi, Baluchi, Immigrants (from India after the 1947 partition of subcontinent) and others like Saraikees, Kashmiri, Memon, and Hazara as well. Over a period of 5 years, NIBD collected venous blood samples (in EDTA) from 466 individuals having at least 1 affected family member known to have β-thalassemia major/HbE- β-thalassemia/ HbE homozygotes/ β-thalassemia trait. Chorionic villus sampling (CVS) at 11 to 15 weeks gestational age for 143 couples referred by thalassemia clinics (for pregnancies at risk of having affected child) was also used to obtain allele information. In all, 648 mutated alleles were identified. The diagnosis of β-thalassemia trait, β-thalassemia major, and Hb E thalassemia were established from clinical data, hematological indices, and hemoglobin electrophoresis (cellulose acetate hemoglobin electrophoresis done by HELENA SAS-MX manual gel tank system), DNA was extracted from whole blood by using Genomic DNA Purification Kit (Gentra system USA). For detection of mutations, a PCR-based method Multiplex ARMS was used, which previously has been used as a mutation-screening tool as mentioned in a number of publications. [9],[10] Primers were designed for simultaneous detection of the following previously-described mutations [7],[8],[10] in a single reaction: IVS 1-5, Fr 8-9, IVS 1-1, Cd-30, Cd-5, Del 619bp, Cd-15, Fr 41- 42, Fr 16, and Cap +1 along with 2 Hb variants: HbS and HbE.
| Results | | |
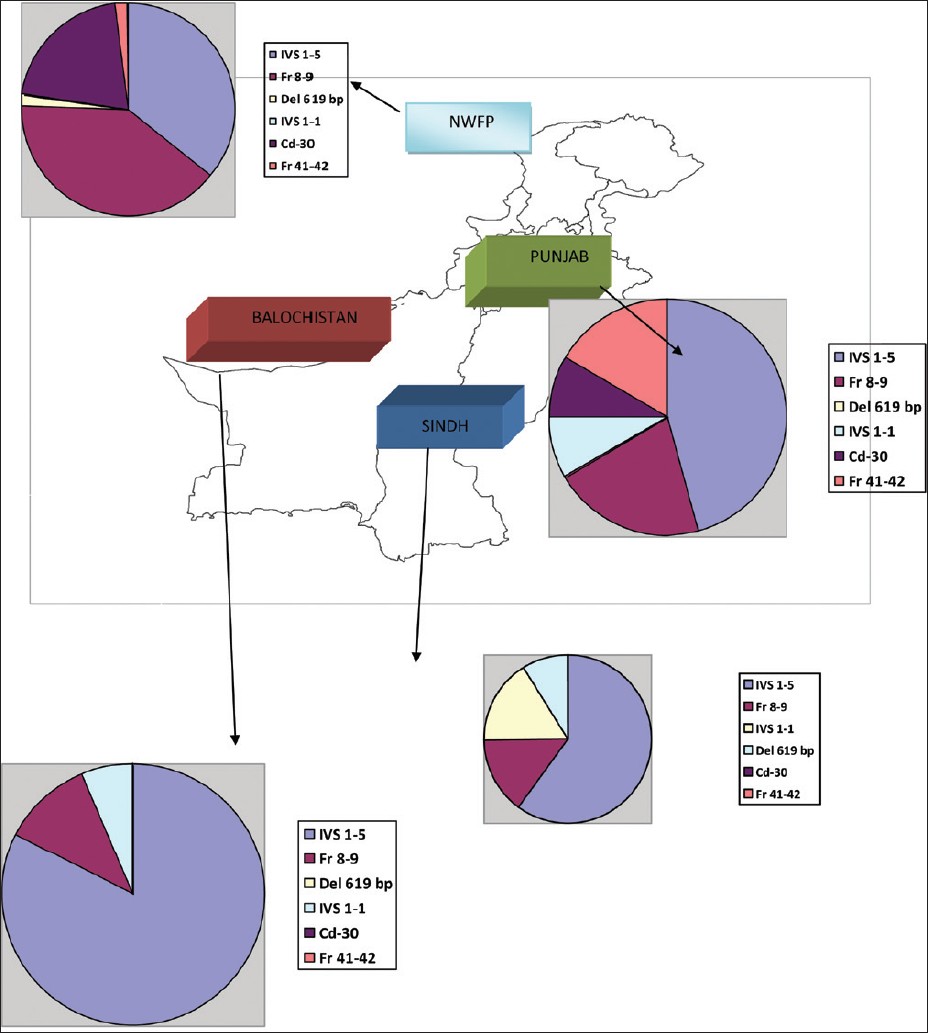
By using the methods mentioned above, mutations were characterized in 640 (98.75%) of the alleles studied; however, there were 8 instances where the allele remained uncharacterized. The study population included 53% males and 47% females. Also, included were 143 CVS specimens, which showed homozygosity in 45 CV samples, whereas 89 of the CV biopsies showed mutations in 1 allele and were diagnosed as carriers; 9 were normal. The overall distribution of the β-thalassemia mutations in different ethnic groups of Pakistan settled in Karachi is summarized in [Table 1]. The genetic heterogeneity in Karachi is reflected by the identification of all the common β-thalassemia alleles and 2 Hb variants, but following 8 mutations were more common: IVS 1-5, Fr 8-9, Del 619 bp, IVS 1-1, Fr 41- 42, Cd-30, Cd-5 and Cd-15, accounting for 93.9% of the β-thalassemia alleles. However, the distribution was uneven as depicted in [Figure 1].{Table 1}
Although IVS 1-5 was the most common mutation (40.89% of the sample), its frequency varied from 20% in the immigrant (from India) population to 76.9% in the Balochis. It should be noted that southwest Iran, which shares border with Balochistan, also has a high prevalence of IVS 1-5. [11] The second most frequent mutation was Fr 8-9, constituting 15.7% of the allele pool. Indeed, Fr 8-9 was the most common mutation in the Pathans (31.3%) as it was in people of Saraikee origin (47%). Higher prevalence of Fr 8-9 in the Pathans has been reported previously also, [7],[8] but other studies [12] have commented that IVS 1-5 is the most common mutation in southern Punjab- the Saraikee belt. This can be attributed to the small count of Saraikee individuals in our study cohort. Deletion 619 bp was found to be the third most common, 11.11% and interestingly, it was identified most commonly (44.6%) in members of the Memon community. The same has been reported by Colah et al.[13] in Gujarat, India as well for the Lohana community (historically, after accepting Islam, the Lohanas were titled as Memons).
| Discussion | | |
Pakistan is a country with a remarkable racial mix from a long history of invasions and commercial interactions, leading to considerable genetic diversity. This supplemented by a strong cultural preference for consanguineous marriage has been responsible for a relatively high prevalence of recessively inherited disorders like β-thalassemia. To sustain children affected with β-thalassemia, monthly blood transfusions accompanied by iron-chelation therapy is needed, and requirements for treating 1 annual birth cohort for 1 year are 90,000 units of blood plus 22 million dollars worth of deferoxamine. [3] Genetic risk factor information such as identification of β-globin gene mutations should be integrated routinely into epidemiological studies, [14] followed by genetic counseling and prenatal diagnosis to reduce birth rate of affected infants [2],[4] to undo the financial burden caused by this disease. We elucidate with this study, the frequencies of different β-thalassemia mutations in the residents of Karachi of various origins. This city is the financial hub of the country, and therefore, people from all regions of Pakistan have migrated over here. Also are included the (post Indo-Pak partition) immigrants from India. The resultant genetic heterogeneity can be seen by the identification of all common β-thalassemia mutations. Results are coherent with statistics provided by Ahmed, et al.[7] in 1996 by characterizing 1216 alleles and also with those given by Khan and Riazuddin[8] who had analyzed 602 β-thalassemia mutations. These investigators narrated a spectrum of 19 mutations and found IVS 1-5 to be the commonest mutation with greater prevalence in Southern Pakistan, whereas Fr 8-9, the second most common mutation, was found more in Northern Pakistan. We also noted that 8 rather than 5 (as previously reported) mutations comprised 93% of the spectrum. Another facet pointed by this analysis is the heterogeneity of genetic spectrum in the immigrant (from India) population residing in Sindh. These people migrated from all parts of the present India at the time of partition of sub-continent and thus represent the same genetic versatility as has been reported by investigators from India, [13] which itself has an ethnically diverse population. Because of the practice of endogamy, gene variants are trapped within extended family groupings and biradris. Lack of a national level screening program and the limited chances of success for changing the social structure on a large scale to avoid consanguineous marriages (which actually does not have a sound ethical footing as well [15] ) emphasize the need for a policy to provide both family studies and premarital or antenatal screening for the relatives of affected children. Knowing the specific mutation panels relevant for the various ethnicities will improve the feasibility of DNA diagnostics in developing countries like ours. Indeed, mutation micro mapping on a caste and biradri basis might serve the purpose even better as differences have been noted in this sub-classification as well. [16] Efforts should be made to increase awareness about the available diagnostic facilities for the prenatal diagnosis in Pakistan.
Based on the outcome of our study, we have formulated a cost-effective proposal for detection of β-thalassemia mutations as shown in [Figure 2]. The strategy is based on first inquiring the ethnic background of the individual with β-thalassemia trait so that DNA can be 'triaged' to the 'common mutation panel' found in the particular ethnic group. If the mutation is not found in the common mutation panel, the DNA is further tested using the uncommon mutation panel. Lastly, it is also worth mentioning that genetic epidemiological studies like ours have more than local relevance- the information can be used in other countries like the USA, Canada, Europe, and Australia in which there is a major influx of Pakistani migrants who present as ethnic minorities for diagnosis, counseling, and management.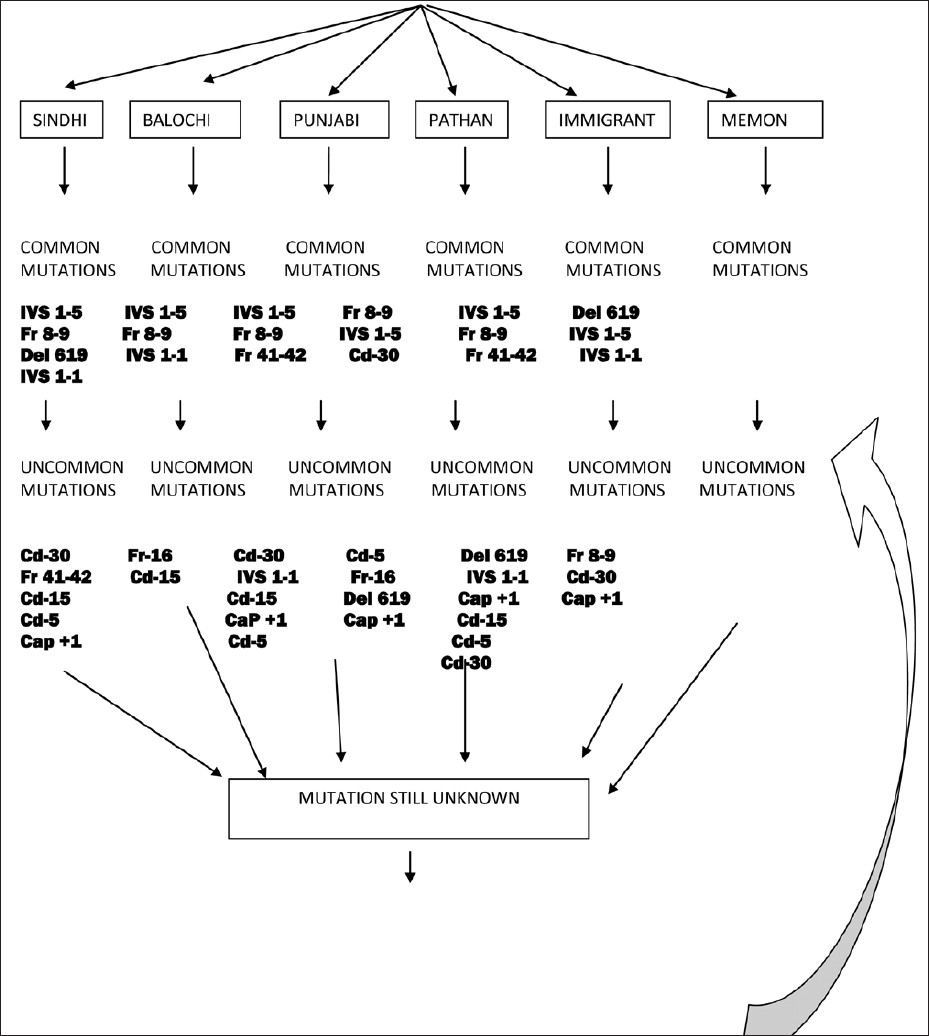 | Figure 2: Individual with β-thalassemia trait based on hematological indices
Click here to view |
Take home message: IVSI-5 (40.89%), Fr8-9 (15.7%), and IVSI-I (8.17%), were the most common genetic mutations identified in Pakistan. Knowledge of the predominant mutation in a given ethnic group will not only help in developing a short panel of (population-specific) primers of mutations, thereby providing a cost-effective method for prenatal diagnosis, but also help the clinicians for genetic counseling and pregnancy termination.
| References | | |
| 1. | Weatherall DJ, Clegg JB. The Thalassemia Syndromes. 4 th ed. Oxford: Blackwell Sci.; 2001. 
|
| 2. | Angastiniotis M, Modell B. Global epidemiology of hemoglobin disorders. Ann N Y Acad Sci 1998;850:251-69.
[PUBMED] |
| 3. | Ahmed S, Saleem M, Modell B, Petrou M. Screening extended families for genetic hemoglobin disorders in Pakistan. N Engl J Med 2002; 347:1162-8.
[PUBMED] |
| 4. | Cao A, Rosatelli MC, Galanello R. Control of β-thalassemia by carrier screening, genetic counselling and prenatal diagnosis: The Sardinian experience. Ciba Found Symp 1996;197:137-51; discussion 151-5.
[PUBMED] |
| 5. | Karimi M, Jamalian N, Yarmohammadi H, Askarnejad A, Afrasiabi A, Hashemi A. Premarital screening for β-thalassemia in Southern Iran: Options for improving the program. J Med Screen 2007;14:62-6.
|
| 6. | Baysal E, Carver MF. The β and δ thalassemia repository. Hemoglobin 1995;19:213-36.
|
| 7. | Ahmed S, Petrou M, Saleem M. Molecular genetics of β-thalassemia in Pakistan: A basis for prenatal diagnosis. Brit J Hematol 1996;94:476-82.
|
| 8. | Khan SN, Riazuddin S. Molecular characterization of β-thalassemia in Pakistan. Hemoglobin 1998;22:333-45.
|
| 9. | Old JM, Varawalla NY, Weatherall DJ. Rapid detection and prenatal diagnosis of β-thalassemia: Studies in Indian and Cypriot populations in the UK. Lancet 1990;336:834-7.
|
| 10. | Ahmed S, Saleem M, Sultana N, Raashid Y, Waqar A, Anwar M, et al. Prenatal diagnosis of β-thalassemia in Pakistan: Experience in a Muslim country. Prenat Diagn 2000;20:378-3.
|
| 11. | Najmabadi H, Karimi-Nejad R, Sahebjam S, Pourfarzad F, Teimourian S, Sahebjam F, et al. The β-thalassemia mutation spectrum in the Iranian population. Hemoglobin 2001;25:285-96.
|
| 12. | Baig SM, Azhar A, Hassan H, Baig JM, Kiyani A, Hameed U, et al. Spectrum of β-thalassemia mutations in various regions of Punjab and Islamabad, Pakistan: Establishment of prenatal diagnosis. Haematologica 2006;91:e13-5.
|
| 13. | Colah R, Gorakshakar A, Nadkarni A, Phanasgaonkar S, Surve R, Sawant P, et al. Regional heterogeneity of β-thalassemia mutations in the multi ethnic Indian population. Blood Cells Mol Dis 2009;42:241-6.
|
| 14. | Shpilberg O, Dorman J, Ferrell R, Trucco M, Shahar A, Kuller LH. The next stage: Molecular epidemiology. J Clin Epidemiol 1997;50:633-8.
|
| 15. | Alwan A, Modell B. Community control of genetic and congenital disorders. EMRO technical publication series 24. Alexandria, Egypt: WHO Regional Office for the Eastern Mediterranean; 1997.
|
| 16. | Hafeez M, Aslam M, Ali A, Rashid Y, Jafri H. Regional and ethnic distribution of β-thalassemia mutations and effect of consanguinity in patients referred for prenatal diagnosis. J Coll Physicians Surg Pak 2007;17:144-7.
|
[Figure 1], [Figure 2]
[Figure 2]
| This article has been cited by | | 1 |
Frequency of G?-globin promoter -158 (C>T) XmnI polymorphism in patients with homozygous/compound heterozygous beta thalassaemia |
|
| Nadir Ali,Muhammad Ayyub,Saleem Ahmed Khan,Suhaib Ahmed,Kazim Abbas,Hamid Saeed Malik,Sunila Tashfeen | | Hematology/Oncology and Stem Cell Therapy. 2015; | | [Pubmed] | [DOI] | | | 2 |
The double helix, a double edged sword: Ethical issues in genetic testing and research |
|
| Anwar, N. | | Journal of Postgraduate Medical Institute. 2013; 27(2): 117-121 | | [Pubmed] | |
|
|
|
|