|
 
 |
|
ORIGINAL ARTICLE |
|
|
|
| Year : 2012 | Volume
: 18
| Issue : 3 | Page : 299-304 |
| |
Analysis of autosomal dominant spinocerebellar ataxia type 1 in an extended family of central India
Shashikant Sharma1, Tekcham Dinesh Singh1, Satish S Poojary1, Manoj Singh Rohilla2, Ajaypal Singh3, Kishore B Lowalekar4, Pramod Kumar Tiwari1
1 Centre for Genomics, Jiwaji University, Gwalior, India
2 Department of Biotechnology, CGO complex, New Delhi, India
3 Department of Medicine, Gajra Raja Medical College, Gwalior, India
4 Department of Neurology, Gajra Raja Medical College, Gwalior, India
| Date of Web Publication | 4-Mar-2013 |
Correspondence Address:
Pramod Kumar Tiwari
Centre for Genomics, Jiwaji University, Gwalior - 474 011
India
 Source of Support: None, Conflict of Interest: None  | 1 |
DOI: 10.4103/0971-6866.107981

 Abstract Abstract | | |
Background: Spinocerebeller ataxia type 1 (SCA1) is a specific type of ataxia among a group of inherited diseases of the central nervous system. In SCA1, genetic defects lead to impairment of specific nerve fibers carrying messages to and from the brain, resulting in the degeneration of the cerebellum, the coordination center of the brain. We investigated 24 members of an extended family in Gwalior city, India, some of which were earlier clinically diagnosed to be suffering from yet unconfirmed type of SCA neurodegenerative disorder.
Materials and Methods: All the family members from each age group were screened clinically and the characteristics of those resembling with ataxia were recorded for diagnosis by MRI. The confirmed patients of the family were genetically tested by PCR based molecular testing to identify the type of SCA (i.e., SCA 1, 2, 3, 4, 6 or 7). Family tree of the disease inheritance was constructed by pedigree based method.
Result and Conclusion: We found the clinical (symptoms and MRI) and genetic (Pedigree and PCR) results to be correlated. The PCR result revealed the disease to be of SCA 1 type being inherited in the family.
Keywords: Cerebellum, magnetic resonance imaging, polymerase chain reaction, pedigree, spinocerebeller ataxia
How to cite this article:
Sharma S, Singh TD, Poojary SS, Rohilla MS, Singh A, Lowalekar KB, Tiwari PK. Analysis of autosomal dominant spinocerebellar ataxia type 1 in an extended family of central India. Indian J Hum Genet 2012;18:299-304 |
How to cite this URL:
Sharma S, Singh TD, Poojary SS, Rohilla MS, Singh A, Lowalekar KB, Tiwari PK. Analysis of autosomal dominant spinocerebellar ataxia type 1 in an extended family of central India. Indian J Hum Genet [serial online] 2012 [cited 2016 Jun 1];18:299-304. Available from: http://www.ijhg.com/text.asp?2012/18/3/299/107981 |
| Introduction | |  |
Spinocerebeller ataxia type 1 (SCA1) is an autosomal-dominant neurodegenerative disease that typically has a mid-life onset, characterized by motor symptoms in the absence of cognitive deficits. [1],[2] In most cases, from the onset of symptoms, the duration of the disease varies from 10-30 years (average 15 years). The onset of symptoms in SCA1 is usually in adulthood, with average age being in the mid-30's (range from <10->60 years). When the onset of symptoms is before age 20, symptoms, in addition to ataxia, occur more frequently. In case of very early onset (before the age of 13) the disease tends to be more severe and progress much more rapidly. SCA1 has the intriguing feature that the disease-causing mutation is the expansion of an unstable tri-nucleotide repeat, specifically a CAG repeat that encodes the amino acid glutamine in ataxin-1gene, located on chromosome locus 6p23. [3],[4],[5],[6],[7] Death usually occurs between 10 to 15 years after the onset of symptoms. The clinical features of SCA1 are highly dependent on the stage of the disease, but typically, in addition to ataxia, include dysarthria and difficulties in swallowing and breathing. At the pathological level, the most frequent and severe alterations seen in SCA1 patients are the loss of Purkinje cells in the cerebellar cortex and the degeneration of neurons in the inferior olivary nuclei, cerebellar dentate nuclei and red nuclei. [2],[6],[8],[9],[10],[11] Numerous observations have established that the polyglutamine repeat, by itself, has a central role in the pathogenesis of polyglutamine diseases, although its effects are strongly modulated by the protein context within which it resides. [12] Nine distinct polyglutamine or triplet repeat disorders and the corresponding genes have, thus far, been identified. [2],[4],[5],[11] The nature of the problematic mutation in these disorders is also the instability in the triplet-repeat tract. [2],[5],[13] For example, in normal Ataxin-1 gene, implicated in Ataxia-1, CAG may be repeated from 6-37 times, but mutant genes are expanded well beyond their normal length, encompassing 40 to more than 100 triplets. [1],[14],[15],[16] Interestingly, the longer the expansion is, the more severe the disease is, and the earlier is the onset.
The Ataxia may be of different types: peripheral, spinal, cerebellar, pseudo-cerebellar, labyrinths and functional ataxia. Overlapping phenotype features of different sub-type make clinical diagnosis difficult. [14] Anticipation, an increase in the clinical severity and younger age of onset of the disease in subsequent generation, is an important feature of autosomal dominant SCA. [17],[18]
In India, the frequency of tri-nucleotide repeat diseases, like spinocerebral ataxia type 2 and 3 is reported to be more common and widely distributed, representing eastern, western, northern and southern regions. [8],[9],[19],[20],[21],[22] The incidence of SCA type 1 is particularly reported from southern populations only. [23] To our knowledge, there is no report on these or related genetic conditions published yet from the central Indian state, Madhya Pradesh, which covers a substantial population size, inhabited by several ethnic populations, including tribes and nomads. Thus, for the first time, we report here about an extended family from Gwalior, MP, clinically diagnosed for inheriting certain type of ataxia. Our detailed analysis revealed SCA type 1 to be inherited in the family, at least for the last four generations, which calls for more extensive screening for SCA in the natives of this region (Central India).
| Materials and Methods | | |
Sample collection
This study was carried out in an extended family, settled in Gwalior, MP, India for the last four generations. A few of the patients in this family were earlier registered in the Department of Neurology, GRMC, Gwalior, for the yet undiagnosed ataxia (SCA) symptoms. Family history data was collected through a questionnaire approved by the institutional ethical committee of GRMC. The neurological assessment was done for all the 27 members of the extended family. Detailed radiological (MRI) examination was carried out at Gajra Raja Medical College (GRMC), Gwalior. Genetic testing for SCA was done in all the family members. After informed written consent, 3-5 ml of blood was taken from adult members of the family and immediately transferred to EDTA coated 10 ml vials. From children, buccal swabs were collected for genomic DNA isolation.
Genomic DNA isolation and PCR amplification
Genomic DNA from peripheral blood lymphocytes or buccal epithelial cells was isolated by standard phenol-chloroform method. [24] The gene sequences for SCA1, SCA2, MJD (SCA3), CACNA1A (SCA6) and SCA7 harboring the CAG-repeat region were amplified in five separate reactions in an Eppendorf Epigradient 96 well thermal cycler (Eppendorf, Germany). The PCR- primers for different genes were adopted from Kumagai et al., (2001) [25] and Dorschner et al., (2002), [26] and got synthesized commercially (Hysel, Germany). PCR reaction mix (25 μl) contained 2.5μl of 10X reaction buffer, 2mM MgCl 2 1μl dNTPs mix (10mM each) (Bangalore Genei, India), 10 pmol of forward and reverse (Hysel, Germany), primers each, 1 unit of Taq polymerase (Hi-Media Pvt Ltd, India) and 50-100 ng genomic DNA. Amplification of the SCA1, SCA2, MJD, and CACNA1A genes were performed for 35 cycles following initial denaturation at 95°C for five minutes, cyclic denaturation at 95°C for one minute, annealing of primers at 61°C for one minute, amplification at 72°C for 1.5 minutes and a final extension at 72°C for five minutes. Amplification of the SCA7 gene was performed following initial denaturation at 95°C for five minutes, cyclic denaturation at 95°C for 45 seconds, annealing of primers at 55°C for 1.15 minutes and 72°C for 60 seconds, with a final extension at 72°C for 10 minutes. Amplified samples were mixed with 0.05% bromophenol blue and electrophoresed through 8% native polyacrylamide gel in 1X TBE buffer with 20bp DNA ladder (Bangalore Genei, India) as size marker. The amplicon were visualized by silver staining [24] and photographed in the UVItek Gel doc system (UK) for further analysis.
| Results | | |
Clinical examination and construction of family tree (pedigree)
The clinical examination of the index case who was admitted to OPD at GRMC, Gwalior, tempted us to screen all the members of his family and the close relatives (daughters and their husband's family). The family (extended) tree (Pedigree) was prepared based on information (physical and clinical) collected from the members of the family through a questionnaire [Figure 1]. Following are the details of the clinical data generated after the examination of different symptomatic and asymptomatic individuals in the family:
- The index case (III-12) was a 24 years old male. Ataxic symptoms appeared two years ago. Ataxic gait dysarthria and other congnitive abilities were normal. His MRI did not show any indication of cerebellar atrophy and fundus examination was normal. A few of his family member were also found affected by similar problem. Ataxia was the main feature in all the patients. Dysarthria, nystamus, spasticity, deep reflexes, and babinski sign were also observed. However, dysmentria, opthalmoplegia, sensory loss, fasiculation, chorea, dementia or mental retardation, epileptic seizures, axonal neuropathy and bladder dysfunction were absent.
- II-2 was paternal aunt of III-12 and was 55 years old. She was totally bed ridden and dependent on others for her routine chores.
- II-4, II-6 and II-13 were paternal aunts. They died due to ataxic problems at the age of 40 yrs, 38 yrs and 25 yrs, respectively. The ataxic features attained severity, which led to their death. Previous medical examination report, if any, could not be obtained.
- II-9 was 40 years old and is the paternal uncle of III-12. He had more severe ataxic features than III-12, unable to perform daily routine work and was wheel chair-bound with ataxic gait, dysarthria, deep reflexes, nystamus, amyotrophy and loss of visual capacity. Fundus examination was normal while MRI showed diffused cerebellar atrophy [Figure 2].
- II-11 was 45 years old and is the paternal uncle of III-12. His ataxic features were mild. He was able to perform all routine work independently. Ataxic gait and visual capacity loss were found.
- III-6 (35 years) and III-7 (25 years) were the sons of II-4. They were asymptomatic patients; however, their walking style was like a drunken person. Some times their speech gets slipped, but they were living normal life.
- III-10 was the son of II-6; he was 32 years old having ataxic gait and dysarthria. CT scan report showed cerebellar atrophy, but fundus examination report was normal. The visual capacity loss was also reported.
- III-2 was son of II-2. He was 35 years old and an asymptomatic patient. The ataxic characters were established a year ago, the speech was slurred, but was able to perform his routine work.
- III-14 (12 years), III-15 (18 years). III-16 (16 years), III-17 (13 years), IV-1 (5 years), IV-2 (8 years), IV-3 (6 years) and IV-4 (4 years), may or may not suffer from this problem in future, which will be known only when they grow older.
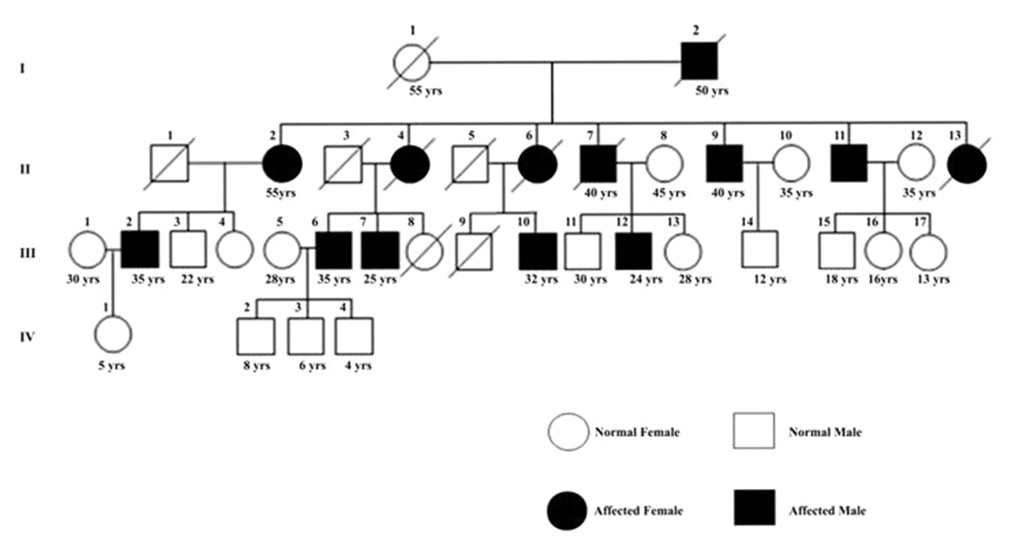 | Figure 1: The family tree (Pedigree) of SCA1 affected Individuals. Numbers below symbols indicate the present age. Filled symbols indicate clinically affected individuals
Click here to view |
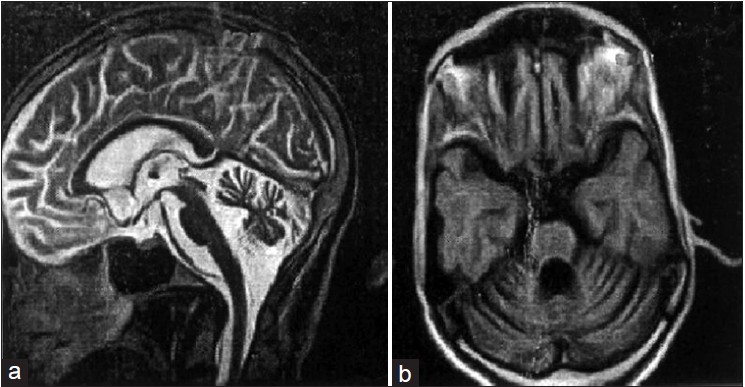 | Figure 2: (a) MRI of 40 years old SCA1 patient II9. Strictly mid-sagittal section, showing normal Fundus and diffused cerebellar atrophy and preserved brainstem, (b) Image on axial section through the pons and the cerebellum. The cerebellar vermis and cortex are atrophied
Click here to view |
All other neurological problems were excluded by different neurological examinations. Evaluation of alcohol consumption, abuse of other substances, thyroid function, vitamin B12 and vitamin E level, serum and urine amino acids level, very long fatty acid chains level and brain's magnetic resonance imaging (MRI), were carried out to ascertain the disease. The family showed autosomal dominant inheritance and anticipation phenomenon. Early onset of the disease occurred in 3 rd generation, with disease symptoms appearing between 22 to 28 years of age, and in the 2 nd generation individuals it was between 36 to 42 years.
PCR based molecular analysis
Genomic PCR was performed with various SCA primers (SCA1, SCA2, MJD, CACNA1A and SCA7) to check which primer set/s amplifies the repeats, if any. Interestingly, only SCA1 primers gave clear amplification with the genomic DNA isolated from different members of the family (both symptomatic as well as asymptomatic) but not others. [Figure 3] shows a representative gel pattern of amplicons (CAG repeats/expansion) for SCA1 obtained from PCR amplification of genomic DNA. We analysed 24 members of the family, of which five were found severely affected. Ataxic feature were diagnosed slowly in other Patients. The PCR-based molecular genetic analysis of all the pedigree members revealed that the normal allele size (bp) was from 150 to 200 bp, while pathological range of expansion was from 210 to 410 bp. Some of the members of the pedigree had allele expanded to more than 230 bp, but did not show ataxic symptoms, which might be due to incomplete pentrance of the gene. Further variation in the severity of spinocerebellar features were also observed in different member of the pedigree in the same generation (age group), which may likely be due to variation in the degree of penetrance of the gene in these pedigree members (Harding 1981). Seven members of the family were below 18 years, but shared an affected Ataxic Gene copy. Gradual loss of vision was reported in all the severely affected patients; however, their Fundus examination did not reveal any visible pathological change. Amyopathy was also observed in two of the patients II 2 and III12. MRI images of the II9, III10 and III12 showed diffused cerebellar atrophy, cerebellar atrophy and normal image, respectively. Both maternal and paternal transmissions were observed in the family. Maternal transmission appeared less severe than paternal transmission of spinocerebellar ataxia. Anticipation phenomenon was clearly seen in the family members. Younger patients were more severely affected than their uncles. Family history suggested that close relative marriages are common in the community to which they belong.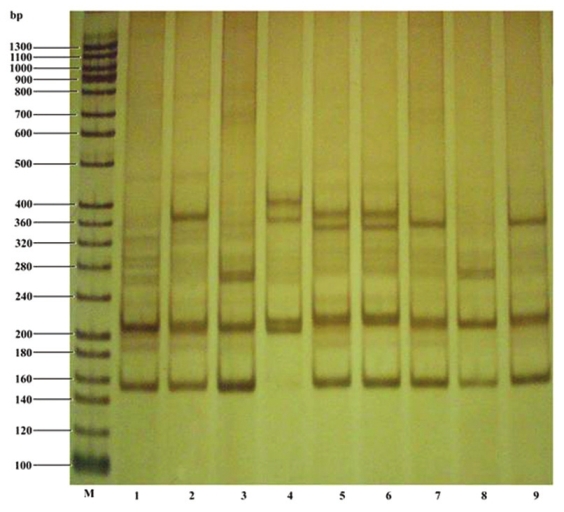 | Figure 3: Silver stained native polyacrylamide gel showing PCR amplified CAG repeats of normal and expanded SCA1 alleles. The lane M represents 20bp ladder as molecular weight or size marker. Lanes 1, 2, 3, 4 and 9 represent expansion of SCA1 allele of affected individuals III12, III2, II11, III10 and II9, respectively. Lane 5, 6, 7, and 8 represent unaffected individuals III11, IV1, II8 and II10, respectively, in the pedigree. The individuals II8 and II10 are women of the family, who come from outside the family
Click here to view |
| Discussion | | |
Significant geographical and ethnic variations are reported in the pattern of distribution of ataxia syndrome in India. A few studies reported the frequency of tri-nucleotide repeat diseases, like Spinocerebrellar ataxia (SCA) type 2 and 3, to be more widely distributed in India than ataxia of other types. [8],[15],[19],[20],[21],[27] The exact reason for such variations is still not clear, but multiple founder mutations are indicated as a possible reason. [14] The incidence of SCA 1 is generally reported from Southern region (Chennai, Tamil Nadu and Bangalore, Karnataka). [14],[23] In community-based studies also, the prevalence rate of ataxia-1 was reported to be 1-2 per 1,00,000 population. [6] To our knowledge, no report has yet been published on the incidence of SCA from central Indian region. The present study is, thus, first in this respect.
Harding (1982) classified autosomal dominant cerebellar ataxia (ADCA) into three types, ADCA I, II and III. [28] ADCA-I is characterized by ataxia with signs of neurodegeneration outside the cerebellum, including pyramidal and extrapyramidal involvements, opthalmoplegia, peripheral neuropathy, and dementia. ADCA-II shows ataxia with extracellular neurologic findings plus retinal degeneration. ADCA-III is the pure form of ataxia, where degeneration is restricted to the cerebellum. SCA 1, 2, 3 and 6 fit into the ADCA-I clinical category. SCA1 is present in 5-27% of all case of SCA and identified by slow, but progressive ataxia, dysarthria, extrapyramidal sign, etc., In the present study, the affected members of the family revealed disease symptoms, including ataxia, dysarthria, nystagmus, spasticity, deep reflexes, balbinski sign and other related symptoms, such as gradual loss of visual capacity. Amyopathy was also observed in two SCA-1 patients II-2 and II-9.
In all types of dominant hereditary SCAs, penetrance is very high in almost every individual having abnormal range of CAG repeats, who is likely to develop symptoms at some time-point in his/her life time, except for a few, who may not develop symptoms ever. There may be significant variation in the age of onset of the disease symptoms as well as the progression of symptoms even within the same family. Individuals having a repeat size between the normal and the expanded range may or may not develop symptoms of SCA, but the offspring of such individuals are likely to be at risk to inherit the progressively increasing repeat length and the occurrence of early disease symptoms (anticipation). [25] This is evident from our present study also, where a few of the family members, (e. g. II8, II10, II12, III2, III3, III11, III13, III15-17 and IV1-2), including women (e. g., II8, II10, II12) from unrelated families, and young children below 10 years of age (e. g., IV1 and IV2) showed the expansion, but not the symptoms. Also, the severity of the symptoms appeared varying in different symptomatic individuals, which might be due to incomplete or no penetrance in those family members or may develop it later. The other likely possibility could be the interruptions caused by CAT repeats within the CAG repeat tracts, which may suppress the disease symptoms. [29] Such a possibility can be detected, if any, by sequencing of amplified fragments from the asymptomatic individuals falling in the range of pathogenic expansion.
Thus, the present study is significant in view of the earlier reports, which showed the incidence of SCA2 and SCA3 to be rather higher and wide-spread in India. While a few reports have indicated the incidence of SCA1 to be more common in southern populations, our present study points this not to be so. Our study strongly supports the view on the presence of multiple founder mutations in Indian populations responsible for distinct geographical and ethnic distribution of SCA types, as suggested earlier. Further, it necessitates intensive screening of the local population or relatives of the families studied in this report for the disease. It might turn out to be more common in this region of the central India and may be due to close family marriages, enforced either by social exclusion of families suffering from such diseases or due to family traditions of small closely-linked communities. Such families need genetic counselling and proper management of the disease to enhance life span and reduce the severity of the disease.
| Acknowledgements | | |
We thankfully acknowledge the Centre for Genomic for material and equipment support to carry out this work through the research grants sanctioned by DBT, DST and ICMR, Govt. of India to Prof. P. K. Tiwari. We are also thankful to the Dean Prof. Sheila Sapre, GRMC, Gwalior, for providing departmental facilities. TDS is a recipient of ICMR-EMR fellowship. We are grateful to the Patient's family member for their cooperation and support in this study.
| References | | |
| 1. | Orr HT. The ins and outs of a polyglutamine neurodegenerative disease: Spinocerebellar ataxia type 1 (SCA1). Neurobiol Dis 2000;7:129-34. 
[PUBMED] |
| 2. | Zoghbi HY, Orr HT. Pathogenic mechanisms of a polyglutamine mediated neurodegenerative disease, spinocerebeller ataxia type 1. J Biol Chem 2009;284:7425-9.
[PUBMED] |
| 3. | Banfi S, Servadio A, Chung MY, Kwiatkowski TJ Jr, McCall AE, Duvick LA, et al. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat Genet 1994;7:513-20.
[PUBMED] |
| 4. | Orr HT, Chung MY, Banfi S, Kwiatkowski TJ Jr, Servadio A, Beaud AL, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 1993;4:221-6.
|
| 5. | Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci 2000;23:217-47.
[PUBMED] |
| 6. | Subramony SH. Disorders of the cerebellum including the degenerative ataxias in Neurology in clinical practice. In: Bradley WG, Daroff RB, Fenichel GM, Jankovic J, editors. 4 th ed. Philadelphia: Butter worth Heinemann; 2004. p. 2169-84.
[PUBMED] |
| 7. | Lorenzetti D, Bohlega S, Zoghbi HY. The expansion of the CAG repeat in ataxin-2 is a frequent cause of autosomal dominant spinocerebellar ataxia. Neurology 1997;49:1009-13.
[PUBMED] |
| 8. | Basu P, Chattopadhyay B, Gangopadhaya, PK. Analysis of CAG repeats in SCA1, SCA2, SCA3, SCA6, SCA7 and DRPLA loci in spinocerebellar ataxia patients and distribution of CAG repeats at the SCA1, SCA2 and SCA6 loci in nine ethnic populations of eastern India. Hum Genet 2000;106:597-604.
|
| 9. | Chakravarthy A, Mukherjee SC. Primary degenerative cerebellar ataxias in ethnic Bengalees in West Bengal: Some observations. Neurol India 2003;51:227-34.
|
| 10. | Gourie-Devi M, Gururaj G, Satishchandra P. Neuroepidemiology in developing countries. 2 nd ed. Bangalore: Prism books; 1997. p. 81-98.
|
| 11. | Orr HT, Zoghbi HY. SCA1 molecular genetics: A history of 13 year collaboration against glutamines. Hum Mo Gene 2001;10:2307-11.
[PUBMED] |
| 12. | Michalik A, Broeckhoven VC. Pathogenesis of polyglutamine disorders: Aggregation revisited. Hum Mole Genet 2003;12:R173-86.
|
| 13. | Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci 2007;30:575-621.
[PUBMED] |
| 14. | Alluri RV, Komandur S, Wagheray A, Chaudhuri JR, Sitajayalakshmi, Meena AK, et al. Molecular Analysis of CAG Repeats at Five Different Spinocerebellar Ataxia loci: Correlation and Alternative Explanations for Disease Pathogenesis. Mol Cells 2007;3:338-42.
|
| 15. | Brandt V, Zoghbi HY. Spinocerebral ataxia type 1. Gene Clinics: Clinical Genetic Information Resource (database online). Seattle: University of Washington; 2001. Available from: http://www.geneclinics.org/profiles/sca1. [Last Accessed on 2001].
|
| 16. | Chung MY, Ranum LP, Duvick LA, Servadio A, Zoghbi HY, Orr HT. Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebeller ataxia type 1. Nat Genet 1993;5:254-8.
[PUBMED] |
| 17. | Sasaki H, Fakazawa T, Yanagihava T. Clinical features and natural history of spinocerebellar ataxia type 1. Act Neurol Scan 1996;9:64-71.
|
| 18. | Svetel M, Culjkovic B, Sternic N, Dragasovic B. Clinico-genetic study of type 1 spinocerebellar ataxia. Svp Arch Celok Lek 1999;127:157-62.
|
| 19. | Kumar S. Spinocerebellar Ataxias: From phenotype to genotype, National Neurosciences Conference Abstracts. Bangalore: NIMHANS; 2004. p. 36-7.
|
| 20. | Saleem Q, Choudhary S, Mukerji M, Bashyam L, Padma MV, Chakarvarthy A, et al. Molecular analysis of autosomal hereditary ataxia in the Indian population: High Frequency of SCA 2 and evidence for a common founder mutation. Hum Gene 2000;106:179-87.
|
| 21. | Sinha KK, Worth PF, Jha DK, Sinha S, Stinton VJ, Davis MB. Autosomal dominant cerebellar ataxia: SCA2 is the most frequent mutation in eastern India. J Neurol Neurosurg Psychiatry 2004;75:448-52.
|
| 22. | Wadia NH. A clinicogenetic analysis of six Indian spinocerebellar ataxia (SCA2) pedigrees. The significance of slow saccades in diagnosis. Brain 1998;121:2341-55.
|
| 23. | Mittal U, Sharma S, Chopra R, Dheeraj K, Pal PK, Srivastava AK. Insights into the mutational history and prevalence of SCA1 in the Indian population through anchored polymorphisms. Hum Genet 2005;118:107-14.
|
| 24. | Sambrook J, Russell DW. Molecular cloning: A laboratory manual, 3 rd ed. New York: Cold Spring Harbor Laboratory Press; 2001.
|
| 25. | Kumagai Y, Sugiura Y, Shimoji S, Kumagai T, Tochikubo S, Yamamoto T. Incidence of Genetic Subgroups of Hereditary Spinocerebellar Ataxia in Fukushima Prefecture. Tohoku J Exp Med 2001;195:85-91.
[PUBMED] |
| 26. | Dorschner MO, Barden D, Stephens K. Diagnosis of Five Spinocerebellar Ataxia Disorders by Multiplex Amplification and Capillary Electrophoresis. J Mol Diagn 2002;4:108-13.
[PUBMED] |
| 27. | Ghosh B, Gangopadhaya PK, Saha S. Genetic study of adult onset inherited progressive ataxia. J Assoc Neuroscientists Eastern India 2000;5:51-4.
|
| 28. | Harding AE. The clinical features and classification of the late onset autosomal dominant cerebellar ataxias. A study of 11 families, including descendants of the Drew family of Walworth. Brain 1982;105:1-28.
[PUBMED] |
| 29. | Krishna N, Mohan S, Yashavanths BS, Ramamurthy A, Kumar HB, Mittal U, et al. SCA 1, SCA 2, and SCA 3/MJD mutations in ataxia syndrome in southern India. Indian J Med Res 2007;126:465-70.
[PUBMED]  |
[Figure 1], [Figure 2], [Figure 3]
| This article has been cited by | | 1 |
Caution Regarding the Interpretation of Homoallelism in Polyglutamine Multiplex Assays |
|
| Danielle C. Smith,Alina Esterhuizen,Jacquie Greenberg | | The Journal of Molecular Diagnostics. 2013; 15(5): 706 | | [Pubmed] | [DOI] | |
|
|
|
|









