BCL-xL Regulates Synaptic Plasticity
Abstract
Mitochondria are the predominant organelle within many presynaptic terminals. During times of high synaptic activity, they affect intracellular calcium homeostasis and provide the energy needed for synaptic vesicle recycling and for the continued operation of membrane ion pumps. Recent discoveries have altered our ideas about the role of mitochondria in the synapse. Mitochondrial localization, morphology, and docking at synaptic sites may indeed alter the kinetics of transmitter release and calcium homeostasis in the presynaptic terminal. In addition, the mitochondrial ion channel BCL-xL, known as a protector against programmed cell death, regulates mitochondrial membrane conductance and bioenergetics in the synapse and can thereby alter synaptic transmitter release and the recycling of pools of synaptic vesicles. BCL-xL, therefore, not only affects the life and death of the cell soma, but its actions in the synapse may underlie the regulation of basic synaptic processes that subtend learning, memory and synaptic development.

Introduction
Transmission of signals through the nervous system requires cell-to-cell communication via neuronal synapses. The basic features of chemical synaptic transmission include close apposition of two nerve cells and release of a chemical neurotransmitter by one cell into the synaptic cleft between two neurons (1). After release, neurotransmitter influences responses of the second neuron via receptors on the postsynaptic cell (2). Neurotransmitter is packaged into small vesicles within the presynaptic terminal, and collections of vesicles wait for a calcium signal produced by calcium entry into the presynaptic cell during action potential firing (3). Elevation of intracellular calcium during synaptic activity enhances the probability of vesicle fusion. Many of the features of synaptic transmission can be enhanced over the short and long term (4). These include changes in presynaptic calcium levels, changes in vesicle numbers and probability of release, and alterations in postsynaptic receptor numbers and function. Such changes lead to short- and long-term modifications in synaptic strength and account in part for plasticity of synaptic activity. Many of these phenomena require energy, and, therefore, may be regulated by mitochondria as will be described in this review. Mitochondria also buffer and re-release calcium inside the synapse, altering the time course and amplitude of the change in calcium concentration during vesicle fusion and recycling (5). Unexpectedly, the BCL-2 family proteins that are known to regulate apoptosis through their actions at mitochondrial membranes have been newly identified as regulators of synaptic activity. Thus, the actions of BCL-xL—a BCL-2 family member—at mitochondria position it to influence learning, memory, and alterations in behavior.
Mitochondria Regulate Synaptic Transmission
Mitochondria are known to be important for synaptic transmission and are the predominant organelle within presynaptic terminals that release neurotransmitter at high rates (6). Mitochondria provide energy in the form of ATP and buffer calcium at these active synapses. Some synaptic mitochondria may buffer calcium even at the expense of ATP production. Indeed, different types of neuronal synapses contain different numbers of mitochondria with slightly different properties, depending on whether the main function of the mitochondria is to provide energy or buffer calcium. At some synapses, oxidative metabolism by mitochondria is crucial to successful neurotransmission, which can be altered considerably––for example, by the rapid onset of synaptic fatigue––if mitochondrial function is eliminated (7). Moreover, mitochondrial bioenergetics are altered acutely in synapses that have undergone conditioning, providing for enhanced oxidative competence (7). Therefore, an interaction may exist between neuronal plasticity and mitochondrial plasticity (8). In this review, we will focus on the role of mitochondria in synaptic transmission and synaptic plasticity and consider possible ways in which the mitochondrial protein BCL-xL brings about changes in mitochondrial properties that may influence these important synaptic events.
Mitochondria Alter Calcium Homeostasis During Synaptic Events
Synaptic transmission depends on mitochondria not only for energy production but also for maintaining calcium homeostasis within the presynaptic terminal (9–15). During synaptic events, calcium influx through voltage-gated channels and the release of calcium from intracellular stores produce elevations of cytosolic calcium that are crucial for synaptic vesicle fusion (16). In the crayfish neuromuscular junction, fast synaptic transmission is dependent on elevated calcium levels inside the presynaptic terminal (10). During a high frequency train of stimuli (tetanus), the amplitude of the response of the post-synaptic cell to neurotransmitter release gradually increases, and even after the tetanus has ceased, the ability of the synapse to release neurotransmitter is increased for up to several minutes (Figure 1A⇓).
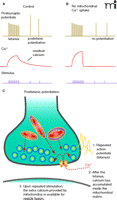
Synaptic potentiation requires mitochondria. A. A diagram of changes in postsynaptic potentials and presynaptic calcium levels during and after tetanic stimulation to the presynaptic cell. B. In the absence of mitochondria, the prolonged tail of residual calcium observed after the tetanus is not apparent and posttetanic potentiation is inhibited. C. Re-release of calcium from mitochondria inside the presynaptic terminal controls short-term synaptic plasticity.
At the very least, although there may be other factors, the ability of the synapse to increase the amount of neurotransmitter released is based on: 1) the ability of the pool of releasable neurotransmitter-containing vesicles to change size, 2) a change in the probability of individual vesicle fusion, or 3) a change the amount of calcium available for release per vesicle (15). Different synapses may have different degrees of potentiation or depression of release of neurotransmitter, both during and after the tetanus, depending on their particular attributes. It has been argued that synapses with a high probability of initial release will depress subsequent release, because they deplete their vesicle pools more rapidly, whereas synapses with a low probability of release will augment release upon increased stimulation, because these synapses contain abundant vesicles that, during baseline stimulation are released less frequently (16).
Although the causes of a change in release probability are complex, both the level of cytosolic calcium and the proximity of sites of calcium influx into the cytosol to sites of vesicle fusion participate in enhancing the probability of fusion events (17, 18). The level of residual calcium during frequent synaptic activity can also play a role in recovery from vesicle depletion (19, 20). In many synapses, tetanic stimulation causes depression of synaptic responses, whereas, as discussed above, in the crayfish neuromuscular junction, neurotransmitter release is enhanced during the tetanus, in part because vesicles may reaccumulate rapidly even during frequent events. Other synapses have different responses to tetanic stimulation. For example, the squid giant presynaptic terminal and the large mammalian central nervous system auditory relay synapse (the calyx of Held) of the medial nucleus of the trapezoid body (MNTB, Box 1) manifest synaptic depression during repetitive stimulation (19, 21). The depression at these synapses is most likely mediated by a high probability of release of vesicles from multiple sites (i.e., active zones) as well as by elevated calcium in the terminals. Under experimental conditions in the squid presynaptic terminal, if extracellular calcium concentration is decreased, then synaptic potentiation can be elicited (21), as in the crayfish synapse.
Characteristics of the MNTB
The medial nucleus of the trapezoid body (MNTB) is located in the auditory brainstem of mammals. It participates in neural pathways that compute the direction of sounds in space by comparing the timing and the intensity of signals that arrive at the two ears. To ensure the accuracy of this information, MNTB neurons, and certain other neurons in these pathways, are capable of firing action potentials at very high rates (600 Hz or more). Such rates are about one order of magnitude faster than most typical neurons (115–118). Moreover a very large presynaptic terminal, termed the calyx of Held, envelops the soma of an MNTB neuron and provides the very strong and secure excitatory input to these cells. These and other features ensure that MNTB neurons fire with very high temporal precision and allow them to lock their action potentials to rapidly changing features of sound stimuli (119, 120). The high energy demands of high frequency activity in both the presynaptic terminals and the postsynaptic cells are associated with mitochondrial specializations, such as the tethering of presynaptic mitochondria directly to the active zones where neurotransmitter is released (36).
Mitochondria participate in shaping the time course and amplitude of neurotransmitter release from presynaptic nerve endings after the invasion of the endings by action potentials. In the example of the crayfish neuromuscular junction, eliminating the ability of mitochondria to sequester calcium during influx through voltage-gated calcium channels leads to a higher rise in intracellular calcium inside the presynaptic terminal during a tetanus but also to prevention of the normal potentiation of neurotransmitter release after the tetanus (Figure 1B⇑) (10). The findings demonstrate that mitochondria are important for the persistent elevation in intracellular calcium (residual calcium) normally found in the presynaptic terminal after it has fired action potentials at a high rate. After mitochondria sequester calcium, they act as a source of persistent release of calcium from the matrix into the cytosol (Figure 1C⇑). In bullfrog sympathetic neurons, mitochondria also slow the rise in intracellular calcium that occurs during a depolarizing stimulus by removing calcium from the cytosol, and slowing the recovery of normal calcium levels after the stimulus (9). At these synapses, mitochondria act as a high capacity buffer of cytosolic calcium and also re-release calcium rapidly in response to a calcium load in the mitochondrial matrix. At the synapse of the MNTB, however, mitochondria play a slightly different role; the effect of mitochondrial calcium sequestration here is to speed the recovery from synaptic depression (20).
Mitochondrial Presence at Presynaptic Sites Regulates Intense Synaptic Activity
We have so far suggested that mitochondria play an important role in regulating neurotransmission in several well-studied models. Another invertebrate model, that of the Drosophila melanogaster neuromuscular junction, provides an ideal system for studying mutations that affect mitochondria and neurotransmission. A genetic screening technique for mutations that affect synaptic transmission in the Drosophila visual system has led to the fascinating finding that multiple genes for mitochondrial targeting are necessary for normal synaptic transmission at the neuromuscular junction (22–24). The first mutated mitochondrial targeting protein to be identified in this screen was Milton (22). Milton binds to kinesin heavy chain, linking mitochondria to microtubules for transport into synaptic endings (25). Animals lacking Milton have abnormal on- and off-transients on electroretinograms, indicating a defect in synaptic transmission to second-order neurons, not in photo-transduction itself (22). Immunoblots and immunocytochemistry performed with Milton-specific antibodies demonstrated that Milton localizes to axonal endings and synaptic sites and is co-localized with mitochondria and with kinesin heavy chain. The mutant photoreceptors contain abundant somatic mitochondria but completely lack synaptic mitochondria. In other ways synaptic morphology is fairly normal. For example, neurotransmitter-containing vesicles are targeted normally to synapses, as evidenced by the presence of synaptic vesicles at active zones, but the density of synaptic vesicles is slightly reduced, suggesting that the lack of mitochondrial targeting influences the establishment or maintenance of vesicle pools in the presynaptic terminal.
Two recent studies have shed further light on the role of mitochondria in vesicle pool dynamics. A genetic screening of Drosophila yielded two other mutants for synaptic transmission, one of which is a mutation in the GTPase dMiro, a protein that participates in the anterograde transport of mitochondria to presynaptic terminals (24, 25). As observed with the Milton mutation, the dMiro-mutated flies lack mitochondria in the presynaptic terminals of neuromuscular junctions. The flies exhibit defects in locomotion and die prematurely. It is fascinating to note that in these dMiro mutants the mitochondria line up in regular rows in the soma and cannot be escorted out to the neuritic processes. The result is a defect in synaptic bouton shape and size and an absence of the normal microtubule loops that form in mature synapses. During high frequency activity at these terminals, there is a slight increase in levels of intracellular calcium compared to controls, and a more rapid fatigue of neurotransmitter release. Calcium is rapidly cleared, however, from the terminals after stimulation has ceased, and this clearance is no different from that of controls. Another striking finding in these synapses is the desynchronization of neurotransmitter release, such that activity causes a barrage of miniature excitatory postsynaptic currents (EPSCs) after the stimuli have ceased. These “minis” are unlikely to be related to calcium homeostasis, which appears to be normal after the stimuli, but may be related to inadequate or delayed functioning of vesicle mobilization inside the presynaptic terminal.
Mitochondrial ATP Production Regulates Normal Functioning Of Synaptic Vesicle Pools
Many studies in synaptic physiology have contributed to the idea that distinct pools of vesicles have different probabilities of release, thoroughly reviewed in Rizzoli and Betz (26). Several different nomenclatures have been employed to describe the pools, but one will be used here (26). The readily releasable pool is defined as the vesicles that are immediately available for release, or “docked” at the active zone. In hippocampal synapses, for example, there appear to be approximately 5–10 vesicles that are docked at each active zone, but a single brief stimulus (such as an action potential) may release only one vesicle. The recycling pool is defined as the pool of vesicles that continue to release and reaccumulate during moderate or physiological stimulation. This pool contains 5–20% of all vesicles, but these estimates vary in different synapses. The reserve pool is defined as those vesicles that only release upon extremely frequent stimulation. The reserve pool of vesicles makes up about 80–90% of the vesicles in most terminals.
Experiments on the temperature-sensitive Drosophila shibire mutant (27) demonstrated that the reserve pool of vesicles is normally mobilized only after the recycling pool is depleted. This mutant exhibits defective endocytosis at high temperatures, leading to an inability of vesicles to re-accumulate after exocytosis. In conditions of mild or moderate stimulation, which would not usually mobilize the reserve pool in controls, the reserve pool is nevertheless mobilized at the high temperatures in the mutant, suggesting that the reserve pool must be used under circumstances where the recycling pool has been depleted. The recycling pool therefore may contain vesicles that are privileged for release, either by their interaction with specific cytoskeletal elements, or their location, or both (28). Surprisingly, however, as observed in synapses where vesicles were fluorescently labeled and then photoconverted for electron microscopy, the recycling pool is not located adjacent to the active zone. Rather, the vesicles of the recycling pool are distributed widely throughout the vesicle cluster (28).
ATP is required for a myriad of cellular processes, and certain steps in synaptic vesicle mobilization, release, and recycling, could be compromised by the lack of locally and rapidly generated ATP. Specific enzyme-dependent steps in synaptic transmission include refilling single vesicles with neurotransmitter (29), membrane fission during endocytosis (30), and coated pit formation (31, 32). Using whole-terminal capacitance measurements of goldfish retinal bipolar neurons, Heidelberger showed that ATP was required for fast compensatory membrane retrieval after exocytosis because dialysis of a non-hydrolyzable form of ATP into the terminal completely and rapidly inhibited endocytosis (30).
Recent evidence suggests that ATP is required for normal functioning of vesicle pools (23). Studies of another mutation in Drosophila that prevents normal synaptic transmission suggest that ATP is needed for mobilizing the reserve pool. In this set of experimental findings, homozygous mutations in the eye for a gene that encodes the dynamin GTPase family mitochondrial fission protein DRP1 (dynamin-related protein 1) caused abnormal synaptic transmission, as evidenced by abnormal electroretinograms. In the mutated flies, mitochondrial movement into presynaptic sites at the photoreceptor synapses was absent, but presynaptic morphology appeared normal in other ways. Photoreceptor somata contained numerous mitochondria that were functional. In the neuromuscular junction, however, mitochondria were conspicuously absent, and resting calcium levels were twice as high as those observed in controls.
Synaptic transmission at the neuromuscular junction failed during intense stimulation, and this effect was temperature-dependent, suggesting that transmission is normally mediated by a metabolic change within the synapses. In addition, the effect on failure of synaptic transmission during intense stimulation was partially rescued by perfusion of ATP into the synapse. Verstrecken et al. reasoned that during intense stimulation, mobilization of vesicles from the reserve pool might require local ATP synthesis. Relying on a previous finding that the recycling pool of vesicles refills constantly during stimulation, but that the reserve pool fills only after stimulation has ceased (27), Verstrecken and colleagues were able to use the styryl dye FM1-43 to differentiate between effects of the mutation on the two different pools (Figure 2A⇓). FM1-43 is taken up into synaptic vesicles during vesicle recycling, where it fluorescently labels collections of vesicles. By stimulating the nerve in a way that produced exocytosis of vesicles from, and endocytosis of vesicles to, the recycling pool alone, the authors demonstrated that there were no differences in the properties of the recycling vesicle pool between the mutants and the controls (Figure 2A, B⇓).
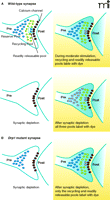
Drosophiladrp1mutation prevents mobilization of neurotransmitter-containing vesicles from the reserve pool. A. Labeling of distinct pools of synaptic vesicles with FM 1-43 in the wild-type synapse is achieved with different stimulation paradigms. B. In the mutant synapse, lack of mobilization of reserve pool vesicles prevents dye uptake into the reserve pool during stimulation as compared to control in A. See text for details.
Although recycling pool endocytosis–exocytosis kinetics appeared to be normal, the endocytosis–exocytosis kinetics of the reserve pool were not. The authors determined that the difference in the mutants was in the ability of the reserve pool to take up dye (Figure 2B⇑). By adding the dye to the bath after strong depletion of all pools, and letting the cells re-accumulate their vesicle pools in the presence of dye, they discovered that the size of the filled pool in controls was much larger than that of the mutants, and, when they unloaded only the recycling pool of vesicles with a brief stimulus, dye remained in the controls, but not in the mutant synapses, suggesting that the mutant synapses contained a poorly functioning reserve pool. The mutants could be rescued by overexpression of the normal DRP1 protein, or by perfusion of ATP into the synapse. Furthermore, the authors found that control reserve pools could be functionally altered by treatment of synapses with inhibitors of mitochondrial function.
Additional experiments enabled the authors to conclude that the defect in the drp1 mutants was in mobilization of vesicles from the reserve pool, not in the size of the reserve pool. They determined that an ATP-sensitive site was an intracellular motor that moved vesicles from pool to pool in an energy-dependent manner. The ATP sensitive motor turned out to be the myosin light chain because: 1) inhibitors of the mysosin light chain kinase caused the same defect in reserve pool cycling in controls as that seen in the mutants, and 2) in the presence of the myosin light chain kinase inhibitor, the reserve pool defect could no longer be rescued by perfusion of ATP into the synapse.
It is clear that mitochondria need to be targeted to the synapse for synaptic transmission to function normally during intense stimulation. Many questions remain, however. For example, what is the mechanism of mitochondrial targeting to the synapse? When a new synaptic connection is made, what is the role of mitochondria? Does mitochondrial fission help target mitochondria to new synaptic sites? What is the signal that a mitochondrion is needed? How does the release of ATP from mitochondria increase at the time it is needed during intense stimulation? What are other ATP-dependent steps in vesicle pool management?
Axonal Targeting of Mitochondria and Their Docking
The axonal transport of mitochondria may be important for targeting of mitochondria to sites of presynaptic activity. Mitochondria appear to move along the axon via cytoskeletal motors and can move in both directions along the axon, as well as remain stationary for prolonged periods of time when they are presumably docked at a site where they are needed (33). Mitochondria in cortical neurons in culture respond to application of the neurotransmitter glutamate by ceasing all movement and changing morphology, suggesting that neuronal activity and elevation of cytosolic calcium concentrations may play a role in mitochondrial docking, as well as in cessation of movement during excitotoxicity (34). Docking can also occur in response to changes that produce axonal growth or in response to intracellular signaling pathways stimulated by the binding of growth factors extracellularly (35). The anterograde movement of mitochondria employs microtubules and kinesin motors, and it appears that different organelles may utilize different adapter proteins to link them to microtubules (35). As stated above, Milton and dMiro are proteins that bind mitochondria to what is likely to be a large microtubule-based complex of proteins––also including the protein syntabulin––involved in movement (25). After traveling along the microtubule, mitochondria arrive at the synapse, where they transfer from microtubules to an actin-based complex that docks the mitochondrion. This complex most likely includes other membrane anchoring proteins as well as actin, but most of those proteins have not yet been identified (33). As seen in electron micrographs, the brainstem auditory synapse of the MNTB (the calyx of Held), which is specialized to release neurotransmitter at extremely high frequency and fidelity, contains a mitochondrial adherens complex. The complex is a collection of filaments that tethers mitochondria very closely to the synapse in a regulated fashion, orienting the matrix cristae perpendicular to the active zone (36). It is likely that the organization of mitochondria within this specialized synapse enables the mitochondria to carry out precisely timed ATP release and calcium buffering. In hippocampal neurons, which have considerably different synaptic organization than that observed in the calyx of Held, it appears that mitochondria are mostly untethered and that they sometimes move and sometimes remain stationary. When hippocampal neurons are stimulated by local application of growth factors to points on the axon, mitochondria move preferentially to the stimulated site, presumably mimicking the in vivo situation where mitochondria might be targeted rapidly during growth or plasticity (37). At synaptic sites, mitochondria bind actin, under the control of phosphatidylinositol-3′ kinase (PI3K) (35, 38).
Function of DRP1 in Synaptic Targeting and Localization
Fusion and fission of mitochondria are dynamic processes that occur within many cell types (39). Whether mitochondria exist as an interconnected network or as individual, discrete organelles most likely depends on the requirements of the individual cell type. The equilibrium between fusion of individual mitochondria and fission of mitochondria into two or several individual mitochondria is a complex and highly regulated process involving the replication and segregation of mitochondrial DNA (40). The proteins that control mitochondrial fission in mammals include Drp1 (41–43) and Fis1 (44). Proteins that control fusion include OPA1 (for Optic Atrophy Type 1, a dynamin-related GTPase) (39), and Mitofusin 1 and 2 (45, 46). During apoptosis, mitochondria fragment under the control of the mitochondrial fission proteins (47, 48), and this fragmentation and some of the features of cell death can be prevented (48, 49) by overexpression of Drp1K38A, a dominant negative mutant of Drp1 (41).
In neurons, a putative function of mitochondrial fission is presumed to create more mitochondria during growth, and particularly to target mitochondria to nascent synapses during development or times of synaptic plasticity. In a study of the role of mitochondrial targeting and fission in the postsynaptic compartment of hippocampal neurons in culture, Li et al. (50) determined that 8–9% of the total cellular mitochondria were found within or close to dendritic protrusions (the site of contact with the presynaptic cell), and that the time of greatest co-localization of mitochondria with dendritic spines was during active phases of synaptic development. At these developmental stages, in resting cells, approximately 10% of dendritic spines contained mitochondria. After repetitive depolarization of the neurons, however, mitochondria changed shape from elongated structures to aggregated clusters and 21% redistributed rapidly to dendritic spines (at three hours after stimulation), suggesting that acute alterations in mitochondrial morphology could play a role in synaptic plasticity. When a stimulating electrode was placed on the cell, mitochondria were found to be more likely to change shape the closer they were to the site of stimulation, and the morphological changes of the mitochondria were prevented by inhibition of NMDA receptors, suggesting that the changes in mitochondrial shape and location were correlated with synaptic excitation. The changes in mitochondrial morphology could also be brought on by overexpression of Drp1 and inhibited by overexpression of Drp1K38A. Accordingly, the number of synapses was increased in neurons overexpressing Drp 1. In contrast, the number of synapses was decreased in controls overexpressing the dominant negative mutant of Drp1K38A, indicating that Drp1 was both required and limiting for the development and plasticity of spines and synapses. Li and colleagues also studied the effect of activity on mitochondrial fission and fusion by time-lapse microscopy. They found that a decrease in neuronal activity in neurons treated with tetrodotoxin (TTX) (which prevents action potential firing) increased the rate of fusion over fission, whereas increased activity in the setting of neuronal depolarization caused an increase in fission over fusion, presumably to make new mitochondria that would be available for new or increasingly active synaptic sites.
Mitochondrial Ion Channels
Specific targeting of mitochondria is thus required for normal synaptic transmission at high frequencies. The regulated targeting of mitochondria to sites of high energy demand suggests that the mechanisms of ATP production and release by mitochondria could very well be regulated during frequent synaptic events. Mitochondria are suggested to release ATP via the voltage-dependent ion channel (VDAC), the most ubiquitous protein in mitochondrial outer membranes. VDAC is the major pathway for the release of metabolites across the mitochondrial outer membrane, and its regulation is important for normal cell function as well as for cell death (51, 52). It is predicted that during synaptic events (such as synaptic plasticity), regulation of the opening of VDAC in the outer mitochondrial membrane could occur. Another prediction is that there is likely to be a second messenger that signals the opening of VDAC during synaptic events.
The first evidence that mitochondrial ion channel activity could be regulated during synaptic events came from studies of mitochondrial membrane conductance during synaptic transmission in an intact presynaptic terminal, that of the squid stellate ganglion. Through the use of a double-barreled patch pipette (53), recordings were made both at rest and during and after intense synaptic stimulation (11).
In control recordings within the resting squid presynaptic terminal, the most frequent mitochondrial ion channel activity was small, with a conductance of less than 50 pS, but other conductances were occasionally seen. In contrast, during frequent electrical stimulation of the squid presynaptic nerve, there occurred a marked increase in activity and conductance of mitochondrial membrane patches within the presynaptic terminal (11). With a delay of less than one second after the onset of nerve stimulation, mitochondrial membrane conductance increased by as much as sixty-fold, a change that persisted for approximately a minute after the stimulus. The delay and persistence of the mitochondrial membrane activity after stimulation implied that the mitochondrial outer membrane channel activity was not simultaneous with the opening of plasma membrane channels and suggested that the increase depended on an intracellular second messenger. Such a messenger could be calcium, which remains elevated in the squid terminal for approximately one minute after stimulation, just as in the crayfish neuromuscular junction and superior cervical ganglion (9, 10, 21). In keeping with these reports, in a calcium-deficient bathing medium, there was no change in mitochondrial conductance in response to stimulation of the presynaptic terminal, demonstrating that the evoked mitochondrial membrane channel activity was dependent on calcium influx (11). Mitochondrial membrane channel activity was also found to be dependent on an intact mitochondrial membrane potential. Uncoupling mitochondria with FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone), completely eliminated the increase in conductance recorded during and after nerve stimulation. The acute changes in mitochondrial membrane conductance were also correlated with synaptic plasticity, because FCCP application also eliminated short term potentiation of the synapse following nerve stimulation.
The possible candidate channels that could be activated on mitochondrial membranes during high frequency activity of the synapse include the channels known to be most abundant in the outer membrane of adult mitochondria in healthy resting neurons such as VDAC (51). The opening of VDAC is most likely very tightly regulated. Kinnally and Tedeschi (54) have pointed out that there are several hundred VDAC channels in a patch that has a diameter of 0.5 μm, assuming a random distribution of channels. If even one channel were open, the patch resistance of a resting mitochondrial membrane would be 1.7 giga-ohms (GΩ) for a channel with a conductance of 650 pS, yet studies have demonstrated the ability to obtain patch resistances of up to 10 GΩ (11, 54). Regulation of VDAC may influence important functions of the synapse such as learning and memory, because knock out mice lacking two of the three known mammalian isoforms of VDAC display abnormalities consistent with the absence of long term potentiation, the electrophysiological correlate of learning found in hippocampal slice recordings (55). Regulation of VDAC opening influences the flux of ATP and other metabolites across the outer mitochondrial membrane (56) and, therefore, could be the conduit for the synchronous release of ATP or calcium during high frequency synaptic events. The opening of VDAC is also modulated by the presence of NADH on the outside of the outer membrane (57–59), suggesting that the metabolic state of the neuron might determine whether VDAC remains closed or opens.
In the squid presynaptic terminal, the activation of mitochondrial channel activity during synaptic transmission is calcium sensitive. Although the recordings were most likely obtained on outer mitochondrial membranes, the only known calcium-sensitive (as opposed to calcium conducting) channel is an inner membrane channel that is activated by elevated calcium concentrations within the mitochondrial matrix. Thus, Ca2+-dependent responses during synaptic stimulation could represent opening of an inner membrane channel whose activity might be linked to the opening of VDAC in the outer membrane (60). A channel spanning two membranes could permit the efflux of calcium (as well as ATP and other ions and metabolites) from the matrix into the cytosol during synaptic potentiation (9, 10).
BCL-xL Is Expressed in Adult Nervous System
Another important set of proteins expressed in the mitochondrial outer membrane that could be regulated during synaptic events is that of the BCL-2 family. The properties of BCL-xL and other BCL-2 family members position them to regulate the processes of synaptic transmission, synaptic plasticity, and synaptic development. BCL-xL is highly expressed in the mammalian nervous system both during development and in adults (61–64), and is localized at least partially to mitochondria (65). During synaptic development, levels of BCL-xL rise in the brain (66) with a similar time course to that that governs the increase in size of presynaptic vesicle clusters (67, 68). In adult brain, only a few BCL-2 proteins continue to be expressed at high levels including the pro-apoptotic protein BID and the anti-apoptotic protein BCL-xL (66). BCL-2 family proteins both cause and prevent cell death, but their precise mechanism of action is still incompletely understood. Properties of these molecules have been widely studied in the hopes of increasing understanding of the complex set of cellular behaviors that occurs during cell death. Some of the characteristics of the molecules that have been uncovered have shed light on their possible function in the synapse. BCL-2 proteins may either protect the synapse from untimely elimination or contribute to its elimination either during development of redundant synapses or in pathological states such as ischemia and neurode-generative diseases. Several of the known properties of BCL-xL may contribute to mitochondrial function in the synapse.
BCL-xL Regulates Apoptosis
Programmed cell death (or apoptosis) plays an important role in the development and throughout the life of many organ systems, including the nervous system (69). In the nervous system, damaged cells or cells not destined for the adult animal are removed (70). Failure of the death program can lead to growth and proliferation of cancer cells, whereas untimely onset of cell death leads to degenerative changes such as found in Alzheimer Disease, and amyotrophic lateral sclerosis (71). In addition, during some pathological insults to the brain such as ischemia or trauma, some cells die immediately, whereas others meet their demise by turning on a programmed death pathway (72, 73).
BCL-2 family proteins regulate the permeabilization of mitochondrial membranes, release of cytochrome c, and eventual activation of caspases, enzymes that support the breakdown of cellular components (74–78). Although it is widely held that anti-apoptotic proteins protect against cell death, and pro-apoptotic molecules kill cells, it is now also firmly acknowledged that anti-apoptotic proteins such as BCL-2 and BCL-xL can be transformed into pro-apoptotic molecules by activation of endogenous proteases (79–82). In addition, some pro-apoptotic molecules serve important pro-survival functions in neurons and in the synapse (83, 84). In their classical role, however, anti-apoptotic molecules such as BCL-xL regulate and prevent cell death in several ways, including binding to pro-apoptotic molecules (85), thereby preventing the effects of the pro-apoptotic molecules on mitochondrial membrane permeability to cytochrome c and other pro-death factors (86–88); increasing the conductance of the outer mitochondrial membrane to metabolites (89); and possibly by directly altering the efficiency of mitochondrial metabolism (90, 91).
In the synapse, the role of both anti-and pro-apoptotic proteins is emerging. Evidence is accumulating that mitochondrial ion channel activity of the BCL-2 family proteins can strengthen or eliminate a synapse during plasticity or degeneration without causing the death of the cell soma (92–95). Therefore, in addition to their role in controlling cell death, BCL-2 family proteins regulate aspects of synaptic physiology even when cell death is not occurring (83, 93, 94).
BCL-xL is an Ion Channel That Regulates Conductance of the Mitochondrial Outer Membrane
BCL-2 family proteins conduct ions when reconstituted into artificial lipid bilayers (86, 96–98). The three-dimensional structure of BCL-xL consists of two central hydrophobic helices surrounded by five amphipathic helices (99). The structure is similar to that of pore-forming bacterial toxins. In lipid vesicles or planar lipid bilayers, the induction of ion channel activity by BCL-xL is related to its known ability to target to, and insert into, lipid membranes. In these artificial membranes, the channel is cation selective at neutral pH, and displays multiple conductances, with a prominent conductance of 276 pS, and several smaller conductance levels. Some of the small conductance channels appear to display typical single channel behavior, whereas the larger conductances have more complex behavior, indicating that multiple proteins could influence the activity of, or constitute, the channel.
A key feature of the ion channel activity of BCL-xL is that it can induce metabolite exchange across mitochondrial membranes (89, 100). In particular, it performs this function in mitochondria from cells that have been exposed to apoptotic stimuli, such as growth factor deprivation. In this pathological setting, BCL-xL appears to protect cells from death by maintaining VDAC in its open configuration despite the pro-apoptotic effect of an early loss of permeability to metabolic substrates.
A surprising dichotomy of the effects of anti-apoptotic molecules is that they enhance the release of ATP and phosphocreatine from mitochondria (101), but prevent the release of cytochrome c (87), and large fluorescent moieties from artificial lipid vesicles (86). How this works is not completely understood, but one possibility is that binding of BCL-xL and BCL-2 to pro-apoptotic molecules may alter the death promoting functions of the pro-apoptotic molecules (85, 88, 102). Therefore, both the channel activity of BCL-xL and its ability to alter the activities of pro-death molecules through protein-protein interactions may comprise the anti-apoptotic functions of BCL-xL.
BCL-xL Produces Mitochondrial Ion Channel Activity within the Presynaptic Terminal
Whether the ion channel activity of BCL-xL might participate in synaptic plasticity and development apart from its role in protection from cell death is now being explored. As a preview to experiments in mammalian neurons, we have studied the effects of BCL-xL on synaptic plasticity in the squid giant presynaptic terminal, a high fidelity, exctitatory, axo-axonal synapse that is critical for the animal’s escape behavior (93). Squid stellate ganglia are immunoreactive by light microscopy for BCL-xL in the large presynaptic terminal fingers. As is typical for mitochondrial staining in large neurons and axons (7, 8), staining is found throughout the axoplasm, but the density of immunoreactivity is greatest close to the plasma membrane, particularly that apposed to the postsynaptic axon. At higher power, striations in BCL-xL staining likely correspond to the location of the spine-like postsynaptic structures that represent contact points between the pre-synaptic terminal and the postsynaptic axon (103, 104). Observation at still higher power reveals a punctate cytoplasmic pattern that co-localizes BCL-xL with a mitochondria-specific dye.
Further evidence for the mitochondrial localization of BCL-xL in squid was obtained by preparation of a purified mitochondrial fraction from the stellate ganglion (105). Immunoblot analysis of this fraction revealed the presence of squid BCL-xL that co-migrated with recombinant human BCL-xL. Also detected in these fractions was the mitochondrial outer membrane protein VDAC1.
Full-length recombinant human BCL-xL protein produces characteristic channel activity with multiple conductances when applied by patch pipette to mitochondrial patches within the living presynaptic terminal (93). Unitary openings of the channel correspond to conductances between 100 pS and 760 pS, and a series of rapid voltage steps to successive potentials reveals current-voltage relations that are linear or very slightly outwardly rectifying.
BCL-xL Enhances Synaptic Transmission
Because BCL-xL induces mitochondrial ion channel activity and induces a change in mitochondrial membrane conductance within the squid presynaptic terminal, BCL-xL might influence the release of calcium or metabolites into the cytosol that could in turn regulate synaptic responses. In support of this hypothesis, injection of recombinant BCL-xL protein into the presynaptic terminal enhances the rate of rise of postsynaptic responses, resulting in an earlier latency for evoked action potentials in the postsynaptic cell as compared to the latency recorded in control synapses (93). Interestingly, injected BCL-xL protein produced potentiation of synaptic transmitter release in both healthy synapses and in those in which transmission had run down (i.e., decreased) to the point that the postsynaptic potential no longer triggered postsynaptic action potentials. Under these conditions, injection of BCL-xL protein into the terminal enhanced the amplitude of the postsynaptic potential, restored suprathreshold responses, and effectively brought the synapse “back to life.”
The time course of enhanced postsynaptic responses after injection of BCL-xL is longer than the changes produced by opening of mitochondrial ion channels during short-term synaptic plasticity. Enhancement of transmission lasts as long as forty-five minutes in some cells, with an average peak response of twenty minutes after injection, suggesting that perhaps endogenous BCL-xL participates in longer lasting changes in synaptic function. The initial recordings of mitochondrial ion channels during synaptic transmission in response to tetanic stimulation (11) suggested that the activation of a calcium-dependent conductance of the outer mitochondrial membrane regulates short term synaptic changes. Although that conductance is clearly activated during normal physiological behaviors of the synapse, its identity is not clear. Activity of BCL-xL could contribute to such changes in permeability of the outer membrane.
BCL-xL Enhances Recovery from Synaptic Depression
In addition to its ability to stimulate neurotransmitter release in an infrequently active synapse (0.03 Hz), injection into the synapse of recombinant BCL-xL protein also enhanced transmitter release from presynaptic terminals stimulated at 2 Hz, a higher frequency that normally produces a significant degree of synaptic depression (21). This finding suggested that, just as calcium buffering by mitochondria alters the recovery from depression in the MNTB (20), BCL-xL may counteract the effects of synaptic depression on the readily releasable pool (93).
Experiments to test the role of BCL-xL in management of vesicle pools demonstrated that recovery of vesicle pools after synaptic depression is, indeed, regulated by BCL-xL. Different stimulus paradigms were employed in order to study the effects of BCL-xL on the kinetics of different vesicle pools. In the first paradigm, stimulation was carried out at 2 Hz before and after the tetanus (Figure 3A⇓). As reported previously (21), during this basal, high rate of stimulation, synaptic depression occurs as the readily releasable pool is depleted. After the depletion, a more reluctantly releasable set of vesicles is used—that of the recycling pool. During the recovery phase following the administration of a tetanus given against the background of continuous 2 Hz stimulation, vesicles do not re-populate all pools, but re-populate only the recycling pool. The time course of the recovery of the recycling pool is rapid, and is not affected by previous injection of BCL-xL.
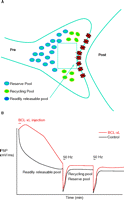
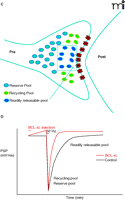
BCL-xL enhances recovery of vesicles to the readily releasable pool. A. During 2 Hz stimulation of the squid synapse, the readily releasable pool remains depleted, and vesicles recycle and re-release from the recycling pool. B. After BCL-xL protein injection into the presynaptic terminal, the postsynaptic potential is enhanced, but after a tetanus, there is no effect on recovery of the recycling or reserve pools. C. Between stimuli at 0.033 Hz, full recovery of all pools occurs. D. Injection of BCL-xL protein into the presynaptic terminal speeds recovery of the readily releasable pool of neurotransmitter. See text for details.
In the second paradigm, recovery from tetanic stimulation was measured during very infrequent basal stimulation (Figure 3B⇑). Under these conditions, full recovery of all pools occurs, as evidenced by the ability of the synapse to release as fully after the tetanus as it does during the control period at the beginning of the experiment. Nevertheless, the time course of recovery of synaptic responses following the tetanus is slower than it is at 2 Hz, suggesting that, when all the pools are re-populated, the most readily releasable––the first pool to be released at the onset of stimulation––re-populates quite slowly (17, 18). The amount of recovery to this pool measured within thirty seconds after the end of the administered tetanus, however, was significantly enhanced by BCL-xL injection when compared to recovery measured in controls. Thus, although synaptic depression during a tetanus is unaffected by BCL-xL, a slow component of the time course of recovery of the total vesicle pool is sensitive to the actions of BCL-xL, and the pool that is affected may be the most readily releasable pool. BCL-xL therefore appears to enhance the ability of a subset of neurotransmitter-containing vesicles to become available for release.
BCL-xL Effects on Synaptic Transmission Do Not Require Calcium Buffering
Calcium release from mitochondria is known to participate in synaptic plasticity; specifically, re-release of calcium from mitochondria following the initial buffering of calcium entering the presynaptic terminal is responsible for the “long tail” of residual calcium that causes posttetanic potentiation at many synapses (Figure 1⇑) (9, 10, 11). At the squid giant synapse in physiological solutions, however, the calcium that enters the terminal during repeated action potentials produces strong synaptic depression, thought to result from depletion of synaptic vesicles (21). Thus, it is unlikely that the enhancement of transmission by BCL-xL, particularly at higher stimulus frequencies (e.g. 2 Hz), results from further elevation of calcium levels alone in the presynaptic terminal.
To examine the potential role of mitochondrial calcium flux in the enhancement of synaptic transmission during BCL-xL injection, neurons were treated with ruthenium red, an agent that is taken up by neurons and inhibits uptake of calcium into mitochondria (106, 107). Ruthenium red blocks short term synaptic potentiation that is dependent upon mitochondrial calcium handling in the synapse. Even under these experimental conditions, however, BCL-xL potentiates transmitter release, suggesting that the actions of BCL-xL in squid presynaptic terminal do not require calcium uptake by mitochondria, and further suggesting that BCL-xL might regulate the local production or release of ATP.
BCL-xL Controls Mitochondrial Bioenergetics
Mitochondria require substrates such as the end products of glycolysis in order to carry out oxidation. Oxidation of substrates hyperpolarizes the mitochondrial membrane potential for the purpose of ATP production. In growing or proliferating cells, growth factors induce cells to increase nutrient uptake from the environment in order to supply the proper amount of substrate for mitochondrial metabolism (91, 108). Nutrients provide energy sources and building blocks for cell growth (91). In the setting of growth factor withdrawal, signals within the cell are activated that can lead to a decrease in the ability of cells to use glycolytic or oxidative substrates. The decline in substrate use eventually causes mitochondrial membrane depolarization. The delicate balance between pro- and anti-apoptotic BCL-2 family proteins appears necessary for the regulation of mitochondrial metabolism at times of deprivation and controls the onset of the eventual release of pro-apoptogenic factors such as cytochrome c into the cytosol (76). Although overexpression of anti-apoptotic BCL-2 proteins such as BCL-xL protects cells from death, in cells that express BCL-xL at normal physiological levels, growth factor withdrawal and metabolic decline can still cause pro-apoptotic proteins to override the protective effects of BCL-xL (91). Whether BCL-2 family proteins participate directly in changes in mitochondrial metabolism in healthy cells is being explored.
Limitation of nutrient stores or oxygen causes the decline in ATP/ADP ratio in the cell cytoplasm. Evidence suggests that, in this setting, BCL-xL acts downstream of metabolic changes in the cell to increase the release of ATP into the cytososl (91). When cells deprived of growth factors were made to overexpress BCL-xL very early in apoptosis, their ability to condense the mitochondrial matrix in response to ADP could be restored, suggesting that, within twelve hours of growth factor deprivation in the absence of BCL-xL, the cause of the change in cellular metabolism is the reversible inability of mitochondria to translocate ADP and ATP across the outer membrane (101). BCL-xL can reverse the pathological situation by activating the opening of VDAC (89). If, despite the protective actions of BCL-xL, the apoptotic program progresses, the eventual release of cytochrome c will occur, and indeed may mark the time of irreversibility of the apoptotic event.
Effects of BCL-xL on Synaptic Transmission Mimicked by Synaptic Perfusion of ATP
If BCL-xL regulates the flux of metabolites across the outer mitochondrial membrane (89, 100), then this property may enhance neurotransmission in the physiological setting. The evidence to support this hypothesis came from studies of the effect of ATP injection into the synapse on the degree of synaptic responses (23, 93). Direct microinjection of ATP into the synapse produced a similar degree and time course of enhancement of synaptic transmission as did the effects of BCL-xL injection (93), and, in fact, occluded the effects of injection of BCL-xL, suggesting that the two agents acted via the same mechanism.
Pro-apoptotic Proteolytic Cleavage Fragment of BCL-xL Causes Synaptic Decline
Growth factor or oxygen withdrawal causes a decline in ATP/ADP ratio in the cell (101). Therefore, it may follow that processes that use a lot of energy such as synaptic vesicle recycling and membrane pumps that maintain ionic homeostasis within the cell are at risk. Mitochondria from growth-factor deprived cells have lost their ability to condense their matrix in response to ADP, and this sign of dysfunction is accompanied by a loss of ability to make ATP during respiration (101).
After the changes in mitochondrial respiration occur, if nutrient or substrate deprivation continues, then apoptotic events at the cell soma may become irreversible. If this occurs in a neuronal synapse, that synapse could be marked for elimination. At this time, a set of changes occurs in the mitochondrial outer membrane that negatively affects synaptic function (75, 105, 109, 110). Under pro-apoptotic conditions, BCL-2 family proteins activate large channel activity that participates in the release of cytochrome c, either in the absence of any change to the properties of the inner membrane (112) or, as may occur during ischemia, accompanying induction of permeability transition of the inner mitochondrial membrane (75).
In the squid synapse, the effects of hypoxia serve as a model to study the role of BCL-xL in neuronal injury (94, 105). The presynaptic terminal is very sensitive to hypoxia, which attenuates synaptic transmission over 10–30 minutes (94). Patch clamp recordings of mitochondrial membrane channel activity during hypoxia revealed large conductance activity not found frequently in controls. The channel activity was larger than that induced by pipette-mediated application of BCL-xL protein.
Injurious stimuli such as hypoxia promote the N-terminal proteolytic cleavage of BCL-xL to form the killer protein ΔN-BCL-xL, which induces cell death and cytochrome c release (79, 80, 81). The large conductance channel activity recorded in the outer mitochondrial membranes of hypoxic synaptic terminals therefore could be a result of activity of proteolytically altered BCL-xL that has formed a new kind of channel activity in the outer mitochondrial membranes. In support of this, when recombinant ΔN BCL-xL protein was added to the patch pipette during mitochondrial recordings within the synapse or to recordings of isolated mammalian brain mitochondria (111) large conductance channels were induced in mitochondrial outer membranes (105, 111). In addition, the appearance of the hypoxia-induced channel in squid could be prevented by pre-treatment of the synapse with zVAD-fmk, a pan-caspase/calpain inhibitor that prevents the cleavage of BCL-xL. Immunoblots confirmed loss of the BCL-xL protein during hypoxia, and although antibodies against the proteolytic cleavage fragment ΔN BCL-xL did not function in squid, in mammalian brain, neurons that had undergone ischemic injury manifested a high level of ΔN BCL-xL in mitochondria (111). Appearance of the channel associated with ΔN BCL-xL during hypoxia most likely arose from specific proteolysis of BCL-xL and not from general injury, because levels of VDAC were preserved in both zVAD-treated and untreated hypoxic synapses, whereas only in the hypoxic synapses treated with zVAD were levels of BCL-xL preserved (94).
ΔN BCL-xL produces loss of membrane potential and cytochrome c release from mammalian mitochondria (80, 113). When ΔN BCL-xL protein was injected into the squid presynaptic terminal, it caused a marked decrease in synaptic responses, the opposite effect of that observed with full-length BCL-xL, even though both variants of recombinant protein produce channel activity when added to the pipette during recordings of mitochondria inside the synapse. The time course of rundown of synaptic responses matched that of hypoxia, suggesting a correlation between the two types of synaptic decline (114). In addition, the data suggested that large mitochondrial channel activity such as that recorded in the setting of hypoxia or with recombinant ΔN 76 BCL-xL could cause the synaptic decline, whereas the smaller conductance changes produced by full length BCL-xL produce synaptic potentiation (84, 114). A further understanding of how the different channel activities produce differential effects on mitochondrial physiology and how those effects, in turn, alter synaptic responses is needed.
VDAC participates in large conductance mitochondrial membrane activity
VDAC is a relatively non-selective channel that is believed to be the major conductance pathway for metabolites such as ATP, ADP, and creatine phosphate across the mitochondrial outer membrane (51, 89, 100). Although BCL-xL causes channel activity in artificial lipid membranes, whether it does so in mitochondrial membranes, or whether it produces all its effects though its biophysical interactions with VDAC is still partly in question. To address this issue more fully, we have taken advantage of the evidence that NADH reduces the conductance of VDAC in mitochondrial membranes (57, 59) but has no effect on the conductance of ΔN BCL-xL in artificial lipid vesicles (105). Therefore, if BCL-xL produces its effects solely by interacting with VDAC, we would be able to inhibit those effects by application of NADH to mitochondrial membranes and to the synapse itself. Indeed, the activity of recombinant ΔN BCL-xL is attenuated by application of NADH to patches of mitochondria inside the synapse, and both the channel activity produced by hypoxia on mitochondrial membranes and the decline in synaptic responses produced by hypoxia were inhibited by application of NADH to the patches or injection of NADH into the presynaptic terminal during synaptic transmission (94, 111). The findings suggest that, during hypoxic-ischemic injury, the activity of ΔN BCL-xL is produced by its interaction with VDAC, further supporting a metabolic role for the channel activity in cell death in injured neurons.
Conclusions and Future Directions
We have painted a picture of BCL-xL as an important regulator of events inside the synapse. It actions position BCL-xL to play an important role in protecting synapses from a decline in function in the setting of injurious stimuli. Not only may BCL-xL serve as a protector, however, it can also become biochemically altered rapidly inside the synapse, and thereby hasten synaptic decline. The models advanced thus far suggest that the two opposite actions of BCL-xL could help balance synaptic function between under-and overactivity, to protect against both synaptic degeneration and excitotoxic death. Furthermore, a protein so integrally related to mitochondrial metabolism inside the synapse could serve as sensor of synaptic activity, to provide for acute and long term changes in the metabolic properties of the synapse necessary for the changes in synaptic efficacy that underlie memory and learning. Amounts of BCL-xL increase during periods of synaptogenesis in mammalian brain (66), thus, in addition to its actions on increasing the availability of ATP acutely for synaptic transmission, BCL-xL may play an important role in axonogenesis and synaptogenesis, for example, by altering the local production of ATP at the synapse during the formation of new vesicle pools, or in targeting and docking mitochondria to synaptic sites during the process of neuronal maturation (Figure 4⇓).
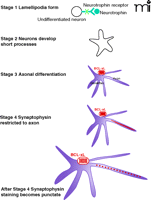
Stages of neuronal growth possibly associated with BCL-xL expression. In the absence of BCL-xL, some neuronal processes and synapses may fail to form or function normally.
BCL-xL is expressed in neurons that are rapidly increasing in size and complexity. The events that occur during neuronal development require not only protein synthesis, but also an ever increasing supply of ATP for energy dependent processes of a neuron of increased size and complexity (Figure 4⇑). If, during neuronal development, BCL-xL increases the efficiency of production of ATP, then it could strongly influence the ability of a neuron to develop to the point of being able to perform the critical functions of rapid and intense release of neurotransmitter that are characteristic of synapses in the adult nervous system. Without crucial changes in mitochondrial morphology, metabolism and targeting, synapses may not form, mature, or display plasticity, because of the absence of local, carefully regulated availability of metabolites.
Acknowledgments
The author thanks J.M. Hardwick for comments and discussion, L.K. Kaczmarek for comments, discussion, and assistance with figure preparation, and J. Eisen for help with manuscript preparation.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Elizabeth A. Jonas, MD, is an Assistant Professor in the Department of Internal Medicine at Yale University School of Medicine. Her laboratory studies how mitochondrial ion channels affect synaptic plasticity. E-mail: elizabeth.jonas{at}yale.edu, fax 203 785-6015.

