Non-nuclear Actions of Estrogen: New Targets for Prevention and Treatment of Cardiovascular Disease
- Karen J. Ho, MD and
- James K. Liao, MD
- The Cardiovascular Division, Department of Medicine, Brigham and Women’s Hospital, and Harvard Medical School, Cambridge,
MA 02139, USA
Abstract
Gender-based differences in the incidence of hypertensive and coronary artery disease, the development of atherosclerosis,
and myocardial remodeling after infarction are attributable to the indirect effect of estrogen on risk factor profiles, such
as cholesterol levels, glucose metabolism, and insulin levels. More recent evidence, however, suggests that activated estrogen
receptor (ER) mediates signaling cascades that culminate in direct protective effects such as vasodilation, inhibition of
response to vessel injury, limiting myocardial injury after infarction, and attenuating cardiac hypertrophy. Although the
ER is usually thought of as a ligand-dependent transcription factor, it can also rapidly mobilize signals at the plasma membrane
and in the cytoplasm. Thus, a greater understanding of ER function and regulation may lead to the development of highly specific
therapeutics that mediate the prevention and treatment of cardiovascular diseases.
INTRODUCTION
Gender-based differences in the incidence of hypertensive and coronary artery disease, the development of atherosclerosis,
and myocardial remodeling after infarction are attributable to the indirect effect of estrogen on risk factor profiles, such
as cholesterol levels, glucose metabolism, and insulin levels (1–3), as well as its direct effects on the myocardium, vascular smooth muscle and endothelium. Although estrogen receptor (ER)
is typically thought of as a ligand-dependent transcription factor, it also modulates the activity of intracellular second
messengers and membrane-associated signaling complexes. In the heart and vasculature, these non-nuclear signaling pathways
mediate rapid vasodilation (4), inhibition of response to vessel injury (5–10), reduction in myocardial injury after infarction (11, 12), and attenuation of cardiac hypertrophy (13, 14).
ESTROGEN RECEPTOR STRUCTURE AND FUNCTION
Both subtypes of ER, ERα and ERβ, are members of the nuclear receptor superfamily (15, 16). They are synthesized from separate genes and are structurally and functionally distinct. Classically, ER regulates gene
expression in target tissues in a ligand-dependent manner: the binding of estradiol (E2) releases ER from an inhibitory complex
and allows for receptor homodimerization and translocation into the nucleus (1, 2, 17). The receptor then binds a palindromic estrogen response element (ERE) located in the promoter region of target genes. The
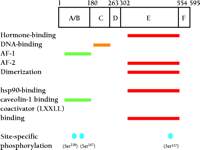
concerted actions of the ligand-independent activation function domain (AF-1) in the N terminus (Figure 1⇓) and the ligand-dependent AF-2 region in the hormone-binding domain lead to the recruitment of tissue-, cell-, and promoter-specific
co-regulator complexes to the ERE, resulting in transactivation or transrepression (18, 19).
Figure 1.
Functional regions of the human estrogen receptor α (ERα). These domains include a ligand-independent transactivation function
domain (AF-1), DNA-binding domain, hormone-binding domain and ligand-dependent transactivation function domain (AF-2). Putative
regions of interaction with other proteins and sites of phosphorylation by various kinases are also shown.
Gene deletion or mutation studies have underlined the importance of ER in cardiovascular physiology (20). Early studies of ovariectomized mice demonstrated that E2 inhibits the proliferation of intimal and medial vascular smooth
muscle (5), suggesting a direct protective effect of estrogen on endothelium and vascular smooth muscle cells (VSMCs). In ERα and ERβ
double-knockout mice, however, E2 inhibits VSMC proliferation but not medial thickening, suggesting that a leakily expressed
splice-variant of ERα could mediate partial protection (21, 22). The more recent production of complete ERα-null mice (23), which exhibit increased medial area, VSMC proliferation, and deposition of proteoglycans in response to vascular injury,
has confirmed the role of ERα in vascular protection (24). The effects also extend to the myocardium. For example, ERα-deficient hearts subjected to whole-organ ischemia and reperfusion
(25) exhibit greater ischemia and higher incidence of arrhythmias than that observed in wild-type hearts. The process may involve
nitric oxide (NO), which ameliorates coronary dysfunction and reduces tissue edema by decreasing microvascular permeability,
because ERα-deficient hearts also demonstrate decreased NO release.
In 1975, Pietras and Szego first described membrane binding sites for estrogen and described a non-genomic mechanism for calcium
influx in endometrial cells (26). More recent studies have added to our current understanding of the highly tissue-specific, non-nuclear ERα signaling network.
Though there is also evidence that ERβ has an important function in the vasculature (27, 28), we focus on ERα because of the greater number of observations that have been made. Defining the cascades through which ERα
elicits its pleiotropic cellular effects and understanding the dysregulation of the network in disease states promises to
uncover novel targets for pharmacological intervention.
NON-NUCLEAR ACTIVITY OF ESTROGEN
Estrogenic transcription-dependent effects, such as those that contribute prominently in organogenesis and function of the
reproductive system, become evident hours after stimulation. Non-nuclear (alternatively referred to as “non-transcriptional”
or “non-genomic”) estrogenic action peaks minutes after stimulation in multiple cell types. Other characteristics include
immunity to inhibitors of DNA transcription or protein synthesis (actinomycin D or cycloheximide) and recruitment of membrane
or cytosol-localized signaling components. These include the second messengers calcium and nitric oxide (NO), receptor tyrosine
kinases including the epidermal growth factor receptor (EGFR) and insulin-like growth factor-1 (IGF-1) receptor (IGF1R), G
protein coupled receptors (GPCRs), and protein kinases including phosphatidylinositol-3' kinase (PI3K), the serine-threonine
kinase Akt, mitogen-activated protein kinase (MAPK) family members, the non-receptor tyrosine kinase Src, and protein kinases
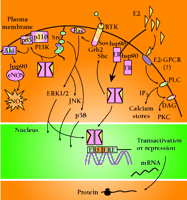
A and C (PKA and PKC, respectively) (Figure 2⇓) (for reviews see 17, 29, 30).
Figure 2.
Selected nuclear and non-nuclear activities of ERα. Details are described in the text. Abbreviations: endothelial nitric oxide
synthase (eNOS), nitric oxide (NO), heat shock protein 90 (HSP90), phosphatidylinositol-3' kinase (PI3K), son of sevenless
(Sos), growth factor receptor binding protein 2 (Grb2), G protein coupled receptor (GPCR), protein kinase A (PKA), protein
kinase C (PKC), extracellular-regulated kinases 1 and 2 (ERK1/2), Jun N-terminal kinase (JNK), 38-kDa isoform of MAPK (p38),
estrogen response element (ERE).
SIGNALING CASCADES ACTIVATED BY ESTROGEN
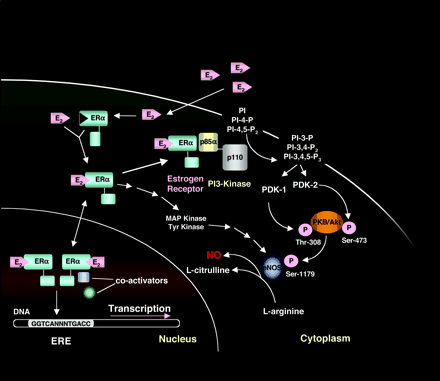
The PI3K-Akt signaling cascade is one downstream target of non-nuclear estrogenic signaling (31–33). In the vasculature, short-term exposure to E2 leads to NO-dependent vasodilation (34). The secretion of NO by healthy vessels relaxes smooth muscle cells and inhibits platelet activation in a cyclic guanosine
3', 5'-monophosphate (cGMP)-dependent mechanism. In cultured endothelial cells, estrogen enhances NO release within minutes
without altering expression of endothelial nitric oxide synthase (eNOS) (33, 35). E2 activates eNOS activity in a biphasic manner through MAPK and PI3K-Akt pathways, leading to enhanced NO release (32). Myocardial protection by high-dose corticosteroids during ischemia-reperfusion injury also appears to be mediated by PI3K-Akt
(36). In both cases, ERα and glucocorticoid receptors activate PI3K by associating with the p85α regulatory subunit in a ligand-dependent
manner (32). Furthermore, the 90-kDa heat shock protein (HSP90) interacts with both eNOS and Akt and modulates eNOS activity by acting
as a scaffold to regulate Akt-dependent phosphorylation of eNOS (37).
MAPK family members are common targets of non-nuclear estrogenic signaling. Induction of eNOS and inducible NOS (iNOS) expression
in cardiac myocytes is blocked by the MAPK inhibitor PD98059 (38). This may be clinically relevant since NO inhibits the activation of caspases and prevents the development of congestive
heart failure (39). Estrogen also activates extracellular-regulated kinases 1 and 2 (ERK1/2) in cardiomyocytes (38), colon cancer (40), breast cancer (41), and bone (42, 43), and inhibits ERK1/2 in VSMCs (44) and lung myofibroblasts (45). In the heart, ERα also selectively activates the 38-kDa isoform of MAPK (p38) to modulate the development of pressure-overload
hypertrophy (13, 14, 46, 47), which is consistent with recruitment of p38 in other models of cardiac hypertrophy (48, 49). In endothelial cells, estrogen prevents disruption of the actin cytoskeleton during ischemia, prevents cell death, and enhances
injury-dependent angiogenesis by rapidly and selectively activating the anti-apoptotic β isoform of p38 (p38β) and inhibiting
pro-apoptotic p38α, leading to the increased expression of MAPK-activated protein kinase-2 (MAPKAP-2) kinase and phosphorylation
of HSP27 (50). Downstream effects include preservation of stress fiber formation and membrane integrity, prevention of hypoxia-induced
apoptosis, and induction of both endothelial cell (EC) migration and the formation of primitive capillary tubes (50).
It is possible that ERα might direct the activation of more receptor-proximal signaling complexes located at the plasma membrane.
When overexpressed in cells, ligand-bound ERα induces the rapid phosphorylation of IGF1R and the activation of ERK1/2. Because
these receptors co-immunoprecipitate in a ligand-dependent manner, a direct physical interaction between ERα and IGF1R could
conceivably mediate the activation of ERK1/2 (51). In breast cancer cell lines, ligand-bound ERα promotes the rapid phosphorylation of the proteins Src and Shc, resulting
in the formation of a Shc-Grb2- (growth factor receptor binding protein 2)-Sos (son of sevenless) complex (52), leading to downstream activation of Ras, Raf, and MAPK. Similarly, in both breast cancer and prostate cancer cells, E2 treatment
induces the association of ERα phospho-Tyr537 with the Src SH2 (Src homology 2) domain, leading to activation of the Src-Ras-ERK pathway and cell cycle progression (53, 54). Additionally, in breast cancer cells, Src modulates PI3K-Akt signaling through a reversible cross-talk mechanism whereby
the ligand-bound ER forms a ternary complex composed of ERα, PI3K, and Src (55). Cross-talk between PI3K and Src has also been observed in osteoclasts and bone marrow cells (56, 57).
Non-nuclear signaling can also amplify the nuclear, transcriptional activity of ERα. For example, in lactotroph cells, E2
rapidly activates ERK1/2, leading to increased transcription of the prolactin (PRL) gene, thus creating an additive effect on PRL expression by complementing the direct ERE-dependent transcriptional activation of PRL by ERα (58). Non-nuclear ERα activity can also elicit ERE-independent transcriptional activation. In cardiac myocytes, E2 rapidly increases
ERK1/2-dependent expression of the early growth response-1 gene (egr-1) by inducing the recruitment of serum response factor (SRF) to serum response elements (SREs) in the egr-1 promoter (59).
Growth factors such as EGF and IGF-1 can stimulate the nuclear activity of ERα through a non-nuclear, E2-independent mechanism.
Through the cross-talk of molecular networks, mitogenic extracellular signals are translated into cell cycle progression or,
in cancer cells, into hormone-independent proliferation (60). EGF- and IGF-1–mediated stimulation of MAP kinases result in the direct phosphorylation of ERα on Ser118 (42, 61, 62), which enhances the binding of p68 RNA helicase (63), and promotes AF-1-dependent transcriptional activity in uterine (64, 65) and ovarian adenocarcinoma cells (66). Nuclear coregulator proteins can also be phosphorylated by ERK1/2 leading to increased transcriptional activity (67). Lastly, Src may enhance AF-1 function of ERα through either a Src, Raf-1, mitogen-activated ERK kinase (MEK) and ERK pathway
that leads to phosphorylation of Ser118, or a pathway that includes Src, MEK kinase (MEKK), Jun N-terminal kinase (JNK) kinase (JNKK), and JNK, and that regulates
AF-1-associated coactivators (68).
MECHANISMS FOR ERα ACTIVITY AT THE PLASMA MEMBRANE
Membrane binding sites for E2 were first implicated in 1977 (26), and additional indirect evidence for a membrane-associated ERα comes from immunohistochemistry (69, 70), overexpressed nuclear receptors (71), or studies with membrane-impermeable ligands (72–74). The trafficking of ERα to different cellular compartments may be regulated by the nature of the stimulation; for example,
in VSMCs transfected with ERα, MAPK activation mediates the nuclear translocation of ERα from the membrane fraction by both
E2-dependent and -independent mechanisms (75). However, because ERα has no intrinsic kinase or phosphatase activity, does not have hydrophobic stretches that could represent
transmembrane domains, and lacks myristoylation and palmitoylation sequences that could anchor it to the membrane, membrane
localization of the receptor seems unlikely. Alternatively, the receptor may associate with membrane caveolae: in fractionated
plasma membranes from endothelial cells (ECs), ERα is localized to caveolae, and E2 stimulates eNOS in isolated caveolae in
an ERα- and calcium-dependent manner (76–78). There is evidence that within the caveolae of ECs, HSP90, eNOS, and cav-1 (caveolin-1, the coat protein of caveolae) exist
in a heterotrimeric complex that modulates eNOS activity depending on intracellular calcium levels (79, 80).
Non-nuclear ERα signaling also involves membrane-associated heterotrimeric G proteins. In Chinese hamster ovary (CHO) cells
transfected with ERα cDNA, treatment of membrane fractions with estrogen activated Gαq and Gαs and rapidly stimulated inositol phosphate production and adenylyl cyclase activity, respectively (71). G protein activation also occurs in ECs, where E2 activation of eNOS can be inhibited with the ER antagonist ICI 182,780,
RGS-4 (a regulator of G protein signaling specific for Gαi and Gαq), or pertussis toxin (specific for Gαi) (81).
POSSIBLE NEW ESTROGEN RECEPTORS
Non-nuclear signaling may involve a receptor altogether distinct from the classical ERα. In macrophage cells, E2 and E2-BSA
induce a rise in intracellular calcium that is inhibitable with pertussis toxin (82, 83). The existence of an E2-GPCR in the hippocampus has also been hypothesized, where E2 stimulation potentiates kainate-induced
currents through modulation of PKA activity (84).
The most notable evidence that estrogen’s non-nuclear effects are mediated by a receptor distinct from ERα or ERβ has come
from studies in the cerebral cortex, where estrogen rapidly stimulates tyrosine phosphorylation of Src, leading to subsequent
Shc–Grb2 complex formation upstream of ERK and B-Raf activation (85, 86). The pathway is not inhibitable by ICI 182,780 in cortical explants from ERα-deficient mice, suggesting that a new receptor,
responsive to E2 but insensitive to ICI 182,780, mediates non-nuclear neuronal differentiation.
The nature of the ERα that mediates the non-nuclear effects of estrogen clearly requires further definition: the distinction
between classical and atypical ERα might be made using cells cultured from complete ERα knockout mice, and ERα-truncated mutants
might provide insight into the specific domains that mediate non-nuclear effects.
NON-NUCLEAR PHARMACOLOGICAL TARGETS
Nonetheless, an increasingly detailed understanding of the ER signaling network and its pleiotropic cellular effects have
made the receptor an attractive pharmacological target. Selective estrogen receptor modulators (SERMs) are ER ligands which
can have varying degrees of agonist or antagonist activities depending on the cell, promoter and coregulator context (87, 88) (Table 1⇓).
Table 1.
Tissue-Specific Effects of Selected SERMs
Tamoxifen, the prototypical SERM, renders indirect cardiovascular protective effects by reducing the amounts of serum total
cholesterol and low-density lipoprotein (LDL) (89). Unfortunately, its strong agonist activity in the endometrium leads to endometrial hyperplasia and low-grade cancers. Raloxifene,
a non-steroidal compound, is similar to tamoxifen but it is less agonistic in the endometrium (90). Though administered primarily for bone preservation, raloxifene also reduces serum triglycerides and serum fibrinogen levels
(91). Like estrogen, raloxifene and its analog LY117018 (92) stimulate eNOS activity in endothelial cells through PI3K- and ERK-dependent pathways, respectively (93), both of which may be involved in coronary artery relaxation (94). Raloxifene also improves endothelium-dependent vasorelaxation in hypertensive rats by enhancing the expression and activity
of NOS (95).
EM-800, a non-steroidal compound, has higher affinity for ERα than any other SERM (96). In addition to demonstrating potent antitumor activity in the uterus and breast, EM-800 may also prevent bone loss and lower
serum cholesterol and triglyceride levels (97). Furthermore, in vitro studies in endothelial cells suggest that EM-800, like E2, enhances NO release by sequential activation
of MAPKs and PI3K-Akt, implicating a direct vascular protective effect (98).
The tissue specificity of SERMs suggests context-specific regulatory mechanisms that depend on the ligand, the promoter of
the target gene, and the combination and exchange of co-regulators (99, 100). Breast cancer and pituitary lactotroph tumors, for example, demonstrate enhanced apoptosis and tumor shrinkage when transfected
with adenovirus constructs containing dominant-negative ERα mutants (101). Because dominant-negative ERα and antiestrogens both recruit transcriptionally repressive proteins to the ERα DNA-binding
complex (102, 103), the co-regulatory proteins that govern ERα activity, in addition to the receptor itself, represent promising therapeutic
targets.
SUMMARY
We are just beginning to appreciate the complexity of ERα signaling. Future research efforts will undoubtedly reveal the intricacies
of expression and translocation of endogenous ERα and possibly the identity of a new receptor that binds E2 and activates
non-nuclear signaling. Furthermore, the activity of coregulators and their role in distinguishing the nuclear and non-nuclear
activities of ERα remain to be defined. A full understanding of these highly cell- and promoter-specific mechanisms will allow
the development of specific agonists and antagonists that selectively elicit only the beneficial effects of estrogen.
Acknowledgments
J.K. Liao is an Established Investigator of the American Heart Association. K.J. Ho is a Howard Hughes Medical Institute Medical
Student Fellow. We thank Dr. A. Senes and S. Gainsbourg for assistance in preparing the manuscript. We apologize to all authors
whose work could not be cited due to space limitations.
- © American Society for Pharmacology and Experimental Theraputics 2002
References
- ↵
Mendelsohn, M.E. and Karas, R.H. The protective effects of estrogen on the cardiovascular system. N. Engl. J. Med. 340,1801–1811 (1999).
- ↵
Babiker, F.A., De Windt , L.J., van Eickels, M., Grohe, C., Meyer, R., and Doevendans, P.A. Estrogenic hormone action in the
heart: Regulatory network and function. Cardiovasc. Res. 53, 709–719 (2002).
- ↵
Stevenson, J.C. Cardiovascular effects of oestrogens. J. Steroid. Biochem. Mol. Biol. 74, 387–393 (2000).
- ↵
White, R.E., Darkow, D.J., and Lang, J.L. Estrogen relaxes coronary arteries by opening BKCa channels through a cGMP-dependent
mechanism. Circ. Res. 77, 936–942 (1995).
- ↵
Sullivan, Jr., T.R., Karas, R.H., Aronovitz, M., Faller, G.T., Ziar, J.P., Smith, J.J., O’Donnell, Jr., T.F., and Mendelsohn,
M.E. Estrogen inhibits the response-to-injury in a mouse carotid artery model. J. Clin. Invest. 96, 2482–2488 (1995).
-
Bourassa, P.A., Milos, P.M., Gaynor, B.J., Breslow, J.L., and Aiello, R.J. Estrogen reduces atherosclerotic lesion development
in apolipoprotein E-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 93, 10022–10027 (1996).
-
Chen, S.J., Li, H., Durand, J., Oparil, S., and Chen, Y.F. Estrogen reduces myointimal proliferation after balloon injury
of rat carotid artery. Circulation 93, 577–584 (1996).
-
Akishita, M., Ouchi, Y., Miyoshi, H., Kozaki, K., Inoue, S., Ishikawa, M., Eto, M., Toba, K., and Orimo, H. Estrogen inhibits
cuff-induced intimal thickening of rat femoral artery: Effects on migration and proliferation of vascular smooth muscle cells.
Atherosclerosis 130:1–10 (1997).
-
Oparil, S., Levine, R.L., Chen, S.J., Durand, J., and Chen, Y.F. Sexually dimorphic response of the balloon-injured rat carotid
artery to hormone treatment. Circulation 95, 1301–1307 (1997).
- ↵
White, C.R., Shelton, J., Chen, S.J., Darley-Usmar, V., Allen, L., Nabors, C., Sanders, P.W., Chen, Y.F., and Oparil, S. Estrogen
restores endothelial cell function in an experimental model of vascular injury. Circulation 96, 1624–1630 (1997).
- ↵
McHugh, N.A., Cook, S.M., Schairer, J.L., Bidgoli, M.M., and Merrill, G.F. Ischemia- and reperfusion-induced ventricular arrhythmias
in dogs: Effects of estrogen. Am. J. Physiol. 268, H2569–H2573 (1995).
- ↵
Node, K., Kitakaze, M., Kosaka, H., Minamino, T., Funaya, H., and Hori, M. Amelioration of ischemia- and reperfusion-induced
myocardial injury by 17β-estradiol: Role of nitric oxide and calcium-activated potassium channels. Circulation 96, 1953–1963 (1997).
- ↵
Farhat, M.Y., Chen, M.F., Bhatti, T., Iqbal, A., Cathapermal, S., and Ramwell, P.W. Protection by oestradiol against the development
of cardiovascular changes associated with monocrotaline pulmonary hypertension in rats. Br. J. Pharmacol. 110, 719–723 (1993).
- ↵
Douglas, P.S., Katz, S.E., Weinberg, E.O., Chen, M.H., Bishop, S.P., and Lorell, B.H. Hypertrophic remodeling: Gender differences
in the early response to left ventricular pressure overload. J. Am. Coll. Cardiol. 32, 1118–1125 (1998).
- ↵
Mangelsdorf, D.J., Thummel, C., Beato, M. et al. The nuclear receptor superfamily: The second decade. Cell 83, 835–839 (1995).
- ↵
Beato, M., Herrlich, P., and Schutz, G. Steroid hormone receptors: Many actors in search of a plot. Cell 83, 851–857 (1995).
- ↵
Hall, J.M., Couse, J.F., and Korach, K.S. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J. Biol. Chem. 276, 36869–36872 (2001).
- ↵
Rosenfeld, M.G., and Glass, C.K. Coregulator codes of transcriptional regulation by nuclear receptors. J. Biol. Chem. 276, 36865–36868 (2001).
- ↵
McDonnell, D.P. and Norris, J.D. Connections and regulation of the human estrogen receptor. Science 296, 1642–1644 (2002).
- ↵
Couse, J.F. and Korach, K.S. Estrogen receptor null mice: What have we learned and where will they lead us? Endocr. Rev. 20, 358–417 (1999).
- ↵
Karas, R.H., Schulten, H., Pare, G., Aronovitz, M.J., Ohlsson, C., Gustafsson, J.A., and Mendelsohn, M.E. Effects of estrogen
on the vascular injury response in estrogen receptor α, β double knockout mice. Circ. Res. 89, 534–539 (2001).
- ↵
Pendaries, C., Darblade, B., Rochaix, P., Krust, A., Chambon, P., Korach, K.S., Bayard, F., and Arnal, J.F. The AF-1 activation-function
of ERα may be dispensable to mediate the effect of estradiol on endothelial NO production in mice. Proc. Natl. Acad. Sci. U.S.A. 99, 2205–2210 (2002).
- ↵
Dupont, S., Krust, A., Gansmuller, A., Dierich, A., Chambon, P., and Mark, M. Effect of single and compound knockouts of estrogen
receptors α (ERα) and β (ERβ) on mouse reproductive phenotypes. Development 127, 4277–4291 (2000).
- ↵
Pare, G., Krust, A., Karas, R.H., Dupont, S., Aronovitz, M., Chambon, P., and Mendelsohn, M.E. Estrogen receptor-α mediates
the protective effects of estrogen against vascular injury. Circ. Res. 90, 1087–1092 (2002).
- ↵
Zhai, P., Eurell, T.E., Cooke, P.S., Lubahn, D.B., and Gross, D.R. Myocardial ischemia-reperfusion injury in estrogen receptor-α
knockout and wild-type mice. Am. J. Physiol. Heart Circ. Physiol. 278, H1640–H647 (2000).
- ↵
Pietras, R.J. and Szego, C.M. Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature 265, 69–72 (1977).
- ↵
Pettersson, K. and Gustafsson, J.A. Role of estrogen receptor β in estrogen action. Annu. Rev. Physiol. 63, 165–192 (2001).
- ↵
Zhu, Y., Bian, Z., Lu, P. et al. Abnormal vascular function and hypertension in mice deficient in estrogen receptor β. Science 295, 505–508 (2002).
- ↵
Collins, P. and Webb, C. Estrogen hits the surface. Nat. Med. 5, 1130–1131 (1999).
- ↵
Moggs, J.G. and Orphanides, G. Estrogen receptors: Orchestrators of pleiotropic cellular responses. EMBO Rep. 2, 775–781 (2001).
- ↵
Honda, K., Sawada, H., Kihara, T., Urushitani, M., Nakamizo, T., Akaike, A., and Shimohama, S. Phosphatidylinositol 3'-kinase
mediates neuroprotection by estrogen in cultured cortical neurons. J. Neurosci. Res. 60, 321–327 (2000).
- ↵
Simoncini, T., Hafezi–Moghadam, A., Brazil, D.P., Ley, K., Chin, W.W., and Liao, J.K. Interaction of oestrogen receptor with
the regulatory subunit of phosphatidylinositol-3'-OH kinase. Nature 407, 538–541 (2000).
- ↵
Hisamoto, K., Ohmichi, M., Kurachi, H., Hayakawa, J., Kanda, Y., Nishio, Y., Adachi, K., Tasaka, K., Miyoshi, E., Fujiwara,
N., Taniguchi, N., and Murata, Y. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular
endothelial cells. J. Biol. Chem. 276, 3459–3467 (2001).
- ↵
Denninger, J.W. and Marletta, M.A. Guanylate cyclase and the NO/cGMP signaling pathway. Biochim. Biophys. Acta 1411, 334–350 (1999).
- ↵
Chen, Z., Yuhanna, I.S., Galcheva-Gargova, Z., Karas, R.H., Mendelsohn, M.E., and Shaul, P.W. Estrogen receptor α mediates
the nongenomic activation of endothelial nitric oxide synthase by estrogen. J. Clin. Invest. 103, 401–406 (1999).
- ↵
Hafezi-Moghadam, A., Simoncini, T., Yang, E., Limbourg, F.P., Plumier, J.C., Rebsamen, M.C., Hsieh, C.M., Chui, D.S., Thomas,
K.L., Prorock, A.J., Laubach, V.E., Moskowitz, M.A., French, B.A., Ley, K., and Liao, J.K. Acute cardiovascular protective
effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat. Med. 8, 473–479 (2002).
- ↵
Fontana, J., Fulton, D., Chen, Y., Fairchild, T.A., McCabe, T.J., Fujita, N., Tsuruo, T., and Sessa, W.C. Domain mapping studies
reveal that the M domain of HSP90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial
nitric oxide synthase and NO release. Circ. Res. 90, 866–873 (2002).
- ↵
Nuedling, S., Kahlert, S., Loebbert, K., Meyer, R., Vetter, H., and Grohe, C. Differential effects of 17β-estradiol on mitogen-activated
protein kinase pathways in rat cardiomyocytes. FEBS Lett. 454, 271–276 (1999).
- ↵
Mital, S., Barbone, A., Addonizio, L.J., Quaegebeur, J.M., Mosca, R.J., Oz, M.C., and Hintze, T.H. Endogenous endothelium-derived
nitric oxide inhibits myocardial caspase activity: Implications for treatment of end-stage heart failure. J. Heart Lung Transplant. 21, 576–585 (2002).
- ↵
Di Domenico, M., Castoria, G., Bilancio, A., Migliaccio, A., and Auricchio, F. Estradiol activation of human colon carcinoma-derived
Caco-2 cell growth. Cancer Res. 56, 4516–4521 (1996).
- ↵
Castoria, G., Barone, M.V., Di Domenico, M., Bilancio, A., Ametrano, D., Migliaccio, A., and Auricchio, F. Non-transcriptional
action of oestradiol and progestin triggers DNA synthesis. EMBO J. 18, 2500–2510 (1999).
- ↵
Endoh, H., Sasaki, H., Maruyama, K., Takeyama, K., Waga, I., Shimizu, T., Kato, S., and Kawashima, H. Rapid activation of
MAP kinase by estrogen in the bone cell line. Biochem. Biophys. Res. Commun. 235, 99–102 (1997).
- ↵
Jessop, H.L., Sjoberg, M., Cheng, M.Z., Zaman, G., Wheeler-Jones, C.P., and Lanyon, L.E. Mechanical strain and estrogen activate
estrogen receptor α in bone cells. J. Bone Miner. Res. 16, 1045–1055 (2001).
- ↵
Hwang, K.C., Lee, K.H., and Jang, Y. Inhibition of MEK1,2/ERK mitogenic pathway by estrogen with antiproliferative properties
in rat aortic smooth muscle cells. J. Steroid Biochem. Mol. Biol. 80, 85–90 (2002).
- ↵
Flores-Delgado, G., Bringas, P., Buckley, S., Anderson, K.D., and Warburton, D. Nongenomic estrogen action in human lung myofibroblasts.
Biochem. Biophys. Res. Commun. 283, 661–667 (2001).
- ↵
Scheuer, J., Malhotra, A., Schaible, T.F., and Capasso, J. Effects of gonadectomy and hormonal replacement on rat hearts.
Circ. Res. 61, 12–19 (1987).
- ↵
van Eickels, M., Grohe, C., Cleutjens, J.P., Janssen, B.J., Wellens, H.J., and Doevendans, P.A. 17β-estradiol attenuates the
development of pressure-overload hypertrophy. Circulation 104, 1419–1423 (2001).
- ↵
Sugden, P.H. and Clerk, A. “Stress-responsive” mitogen-activated protein kinases (c-Jun N-terminal kinases and p38 mitogen-activated
protein kinases) in the myocardium. Circ. Res. 83, 345–352 (1998).
- ↵
Clerk, A., Michael, A., and Sugden, P.H. Stimulation of the p38 mitogen-activated protein kinase pathway in neonatal rat ventricular
myocytes by the G protein-coupled receptor agonists, endothelin-1 and phenylephrine: A role in cardiac myocyte hypertrophy?
J Cell Biol 142, 523–535 (1998).
- ↵
Razandi, M., Pedram, A., and Levin, E.R. Estrogen signals to the preservation of endothelial cell form and function. J. Biol. Chem. 275, 38540–38546 (2000).
- ↵
Kahlert, S., Nuedling, S., van Eickels, M., Vetter, H., Meyer, R., and Grohe, C. Estrogen receptor α rapidly activates the
IGF-1 receptor pathway. J. Biol. Chem. 275, 18447–18453 (2000).
- ↵
Song, R.X., McPherson, R.A., Adam, L., Bao, Y., Shupnik, M., Kumar, R., and Santen, R.J. Linkage of rapid estrogen action
to MAPK activation by ERα–Shc association and Shc pathway activation. Mol. Endocrinol. 16, 116–127 (2002).
- ↵
Migliaccio, A., Di Domenico, M., Castoria, G., de Falco, A., Bontempo, P., Nola, E., and Auricchio, F. Tyrosine kinase/p21ras/MAP-kinase
pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 15, 1292–1300 (1996).
- ↵
Migliaccio, A., Castoria, G., Di Domenico, M., de Falco, A., Bilancio, A., Lombardi, M., Barone, M.V., Ametrano, D., Zannini,
M.S., Abbondanza, C., and Auricchio, F. Steroid-induced androgen receptor-oestradiol receptor β-Src complex triggers prostate
cancer cell proliferation. EMBO J. 19, 5406–5417 (2000).
- ↵
Castoria, G., Migliaccio, A., Bilancio, A., Di Domenico, M., de Falco, A., Lombardi, M., Fiorentino, R., Varricchio, L., Barone,
M.V., and Auricchio, F. PI3'-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 20, 6050–6059 (2001).
- ↵
Wong, B.R., Besser, D., Kim, N., Arron, J.R., Vologodskaia, M., Hanafusa, H., and Choi, Y. TRANCE, a TNF family member, activates
Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol. Cell 4, 1041–1049 (1999).
- ↵
Kubota, Y., Tanaka, T., Kitanaka, A., Ohnishi, H., Okutani, Y., Waki, M., Ishida, T., and Kamano, H. Src transduces erythropoietin-induced
differentiation signals through phosphatidylinositol 3'-kinase. EMBO J. 20, 5666–5677 (2001).
- ↵
Watters, J.J., Chun, T.Y., Kim, Y.N., Bertics, P.J., and Gorski, J. Estrogen modulation of prolactin gene expression requires
an intact mitogen-activated protein kinase signal transduction pathway in cultured rat pituitary cells. Mol. Endocrinol. 14, 1872–1881 (2000).
- ↵
de Jager, T., Pelzer, T., Muller-Botz, S., Imam, A., Muck, J., and Neyses, L. Mechanisms of estrogen receptor action in the
myocardium. Rapid gene activation via the ERK1/2 pathway and serum response elements. J. Biol. Chem. 276, 27873–27880 (2001).
- ↵
Kato, S., Masuhiro, Y., Watanabe, M., Kobayashi, Y., Takeyama, K.I., Endoh, H., and Yanagisawa, J. Molecular mechanism of
a cross-talk between oestrogen and growth factor signalling pathways. Genes Cells 5, 593–601 (2000).
- ↵
Bunone, G., Briand, P.A., Miksicek, R.J., and Picard, D. Activation of the unliganded estrogen receptor by EGF involves the
MAP kinase pathway and direct phosphorylation. EMBO J. 15, 2174–2183 (1996).
- ↵
Kato, S., Endoh, H., Masuhiro, Y. et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated
protein kinase. Science 270, 1491–1494 (1995).
- ↵
Endoh, H., Maruyama, K., Masuhiro, Y. et al. Purification and identification of p68 RNA helicase acting as a transcriptional
coactivator specific for the activation function 1 of human estrogen receptor α. Mol. Cell. Biol. 19, 5363–5372 (1999).
- ↵
Aronica, S.M. and Katzenellenbogen, B.S. Stimulation of estrogen receptor-mediated transcription and alteration in the phosphorylation
state of the rat uterine estrogen receptor by estrogen, cyclic adenosine monophosphate, and insulin-like growth factor-I.
Mol. Endocrinol. 7, 743–752 (1993).
- ↵
Ignar-Trowbridge, D.M., Teng, C.T., Ross, K.A., Parker, M.G., Korach, K.S., and McLachlan, J.A. Peptide growth factors elicit
estrogen receptor-dependent transcriptional activation of an estrogen-responsive element. Mol. Endocrinol. 7, 992–998 (1993).
- ↵
Ignar-Trowbridge, D.M., Pimentel, M., Parker, M.G., McLachlan, J.A., and Korach, K.S. Peptide growth factor cross-talk with
the estrogen receptor requires the A/B domain and occurs independently of protein kinase C or estradiol. Endocrinology 137, 1735–1744 (1996).
- ↵
Rowan, B.G., Weigel, N.L., and O’Malley, B.W. Phosphorylation of steroid receptor coactivator-1. Identification of the phosphorylation
sites and phosphorylation through the mitogen-activated protein kinase pathway. J. Biol. Chem. 275, 4475–4483 (2000).
- ↵
Feng, W., Webb, P., Nguyen, P., Liu, X., Li, J., Karin, M., and Kushner, P.J. Potentiation of estrogen receptor activation
function 1 (AF-1) by Src/JNK through a serine118-independent pathway. Mol. Endocrinol. 15, 32–45 (2001).
- ↵
Pappas, T.C., Gametchu, B., and Watson, C.S. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand
binding. FASEB J. 9, 404–410 (1995).
- ↵
Ropero, A.B., Soria, B., and Nadal, A. A nonclassical estrogen membrane receptor triggers rapid differential actions in the
endocrine pancreas. Mol. Endocrinol. 16, 497–505 (2002).
- ↵
Razandi, M., Pedram, A., Greene, G.L., and Levin, E.R. Cell membrane and nuclear estrogen receptors (ERs) originate from a
single transcript: Studies of ERα and ERβ expressed in Chinese hamster ovary cells. Mol. Endocrinol. 13, 307–319 (1999).
- ↵
Kelly, M.J. and Levin, E.R. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol. Metab. 12, 152–156 (2001).
-
Kuroki, Y., Fukushima, K., Kanda, Y., Mizuno, K., and Watanabe, Y. Putative membrane-bound estrogen receptors possibly stimulate
mitogen-activated protein kinase in the rat hippocampus. Eur. J. Pharmacol. 400, 205–209 (2000).
- ↵
Morey, A.K., Razandi, M., Pedram, A., Hu, R.M., Prins, B.A., and Levin, E.R. Oestrogen and progesterone inhibit the stimulated
production of endothelin-1. Biochem. J. 330, 1097–1105 (1998).
- ↵
Lu, Q., Ebling, H., Mittler, J., Baur, W.E., and Karas, R.H. MAP kinase mediates growth factor-induced nuclear translocation
of estrogen receptor α. FEBS Lett. 516, 1–8 (2002).
- ↵
Chambliss, K.L. and Shaul, P.W. Rapid activation of endothelial NO synthase by estrogen: Evidence for a steroid receptor fast-action
complex (SRFC) in caveolae. Steroids 67, 413–419 (2002).
-
Kim, H.P., Lee, J.Y., Jeong, J.K., Bae, S.W., Lee, H.K., and Jo, I. Nongenomic stimulation of nitric oxide release by estrogen
is mediated by estrogen receptor α localized in caveolae. Biochem. Biophys. Res. Commun. 263, 257–262 (1999).
- ↵
Chambliss, K.L., Yuhanna, I.S., Mineo, C., Liu, P., German, Z., Sherman, T.S., Mendelsohn, M.E., Anderson, R.G., and Shaul,
P.W. Estrogen receptor α and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae.
Circ. Res. 87, E44–E52 (2000).
- ↵
Feron, O., Saldana, F., Michel, J.B., and Michel, T. The endothelial nitric-oxide-synthase–caveolin regulatory cycle. J. Biol. Chem. 273, 3125–3128 (1998).
- ↵
Gratton, J.P., Fontana, J., O’Connor, D.S., Garcia-Cardena, G., McCabe, T.J., and Sessa, W.C. Reconstitution of an endothelial
nitric–oxide synthase (eNOS), HSP90, and caveolin-1 complex in vitro. Evidence that HSP90 facilitates calmodulin stimulated
displacement of eNOS from caveolin-1. J. Biol. Chem. 275, 22268–22272 (2000).
- ↵
Wyckoff, M.H., Chambliss, K.L., Mineo, C., Yuhanna, I.S., Mendelsohn, M.E., Mumby, S.M., and Shaul, P.W. Plasma membrane estrogen
receptors are coupled to endothelial nitric-oxide synthase through Gαi. J. Biol. Chem. 276, 27071–27076 (2001).
- ↵
Benten, W.P., Stephan, C., Lieberherr, M., and Wunderlich, F. Estradiol signaling via sequestrable surface receptors. Endocrinology 142, 1669–1677 (2001).
- ↵
Guo, Z., Krucken, J., Benten, W.P., and Wunderlich, F. Estradiol–induced nongenomic calcium signaling regulates genotropic
signaling in macrophages. J. Biol. Chem. 277, 7044–7050 (2002).
- ↵
Moss, R.L. and Gu, Q. Estrogen: Mechanisms for a rapid action in CA1 hippocampal neurons. Steroids 64, 14–21 (1999).
- ↵
Nethrapalli, I.S., Singh, M., Guan, X., Guo, Q., Lubahn, D.B., Korach, K.S., and Toran-Allerand, C.D. Estradiol (E2) elicits
SRC phosphorylation in the mouse neocortex: The initial event in E2 activation of the MAPK cascade? Endocrinology 142, 5145–5148 (2001).
- ↵
Singh, M., Setalo, Jr., G., Guan, X., Frail, D.E., and Toran-Allerand, C.D. Estrogen-induced activation of the mitogen-activated
protein kinase cascade in the cerebral cortex of estrogen receptor-α knock-out mice. J. Neurosci. 20, 1694–1700 (2000).
- ↵
Osborne, C.K., Zhao, H., and Fuqua, S.A. Selective estrogen receptor modulators: Structure, function, and clinical use. J. Clin. Oncol. 18, 3172–3186 (2000).
- ↵
Burger, H.G. Selective oestrogen receptor modulators. Horm. Res. 53 Suppl 3:25–29 (2000).
- ↵
Williams, J.K., Wagner, J.D., Li, Z., Golden, D.L., and Adams, M.R. Tamoxifen inhibits arterial accumulation of LDL degradation
products and progression of coronary artery atherosclerosis in monkeys. Arterioscler. Thromb. Vasc. Biol. 17, 403–408 (1997).
- ↵
Dardes, R.C., Schafer, J.M., Pearce, S.T., Osipo, C., Chen, B., and Jordan, V.C. Regulation of estrogen target genes and growth
by selective estrogen-receptor modulators in endometrial cancer cells. Gynecol. Oncol. 85, 498–506 (2002).
- ↵
Walsh, B.W. The effects of estrogen and selective estrogen receptor modulators on cardiovascular risk factors. Ann. N.Y. Acad. Sci. 949, 163–167 (2001).
- ↵
Hisamoto, K., Ohmichi, M., Kanda, Y., et al. Induction of endothelial nitric-oxide synthase phosphorylation by the raloxifene
analog LY117018 is differentially mediated by Akt and extracellular signal-regulated protein kinase in vascular endothelial
cells. J. Biol. Chem. 276, 47642–47649 (2001).
- ↵
Simoncini, T., Genazzani, A.R., and Liao, J.K. Nongenomic mechanisms of endothelial nitric oxide synthase activation by the
selective estrogen receptor modulator raloxifene. Circulation 105, 1368–1373 (2002).
- ↵
Figtree, G.A., Lu, Y., Webb, C.M., and Collins, P. Raloxifene acutely relaxes rabbit coronary arteries in vitro by an estrogen
receptor-dependent and nitric-oxide-dependent mechanism. Circulation 100,1095–1101 (1999).
- ↵
Wassmann, S., Laufs, U., Stamenkovic, D., Linz, W., Stasch, J.P., Ahlbory, K., Rosen, R., Bohm, M., and Nickenig, G. Raloxifene
improves endothelial dysfunction in hypertension by reduced oxidative stress and enhanced nitric oxide production. Circulation 105, 2083–2091 (2002).
- ↵
Martel, C., Provencher, L., Li, X., St. Pierre, A., Leblanc, G., Gauthier, S., Merand, Y., and Labrie, F. Binding characteristics
of novel nonsteroidal antiestrogens to the rat uterine estrogen receptors. J. Steroid Biochem. Mol. Biol. 64, 199–205 (1998).
- ↵
Labrie, F., Labrie, C., Belanger, A., Simard, J., Giguere, V., Tremblay, A., and Tremblay, G. EM–652 (SCH57068), a pure SERM
having complete antiestrogenic activity in the mammary gland and endometrium. J. Steroid Biochem. Mol. Biol. 79, 213–225 (2001).
- ↵
Simoncini, T., Varone, G., Fornari, L., Mannella, P., Luisi, M., Labrie, F., and Genazzani, A.R. Genomic and nongenomic mechanisms
of nitric oxide synthesis induction in human endothelial cells by a fourth-generation selective estrogen receptor modulator.
Endocrinology 143, 2052–2061 (2002).
- ↵
Katzenellenbogen, B.S., Choi, I., Delage-Mourroux, R., Ediger, T.R., Martini, P.G., Montano, M., Sun, J., Weis, K., and Katzenellenbogen,
J.A. Molecular mechanisms of estrogen action: Selective ligands and receptor pharmacology. J. Steroid Biochem. Mol. Biol. 74, 279–285 (2000).
- ↵
McDonnell, D.P., Chang, C.Y., and Norris, J.D. Capitalizing on the complexities of estrogen receptor pharmacology in the quest
for the perfect SERM. Ann. N.Y. Acad. Sci. 949, 16–35 (2001).
- ↵
Lee, E.J., Jakacka, M., Duan, W.R., Chien, P.Y., Martinson, F., Gehm, B.D., and Jameson, J.L. Adenovirus-directed expression
of dominant negative estrogen receptor induces apoptosis in breast cancer cells and regression of tumors in nude mice. Mol. Med. 7, 773–782 (2001).
- ↵
Brzozowski, A.M., Pike, A.C., Dauter, Z., Hubbard, R.E., Bonn, T., Engstrom, O., Ohman, L., Greene, G.L., Gustafsson. J.A.,
and Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 389, 753–758 (1997).
- ↵
Montano, M.M., Ekena, K., Delage-Mourroux, R., Chang, W., Martini, P., and Katzenellenbogen. B.S. An estrogen receptor-selective
coregulator that potentiates the effectiveness of antiestrogens and represses the activity of estrogens. Proc. Natl. Acad. Sci. U.S.A. 96, 6947–6952 (1999).
James K. Liao, MD, (left) is the Director of the Vascular Medicine Research Unit at the Brigham and Women’s Hospital, and is a Associate Professor
of Medicine at Harvard Medical School. Address comments to JKL. E-mail jliao{at}rics.bwh.harvard.edu. Fax 617-768-8425 Karen J. Ho, MD, (right) is a member of the Vascular Medicine Research Unit, Brigham and Women’s Hospital and Harvard Medical School.