
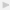

Prediction of Disease Severity in Patients with Early Rheumatoid Arthritis by Gene Expression Profiling
- Zheng Liu zheng.liu{at}mail.utexas.edu1,2
- Tuulikki Sokka tuulikki.sokka{at}ksshp.fi1,3
- Kevin Maas khmaas{at}stanford.edu1,2
- Nancy J. Olsen nancy.olsen{at}utsouthwestern.edu4
- Thomas M. Aune tom.aune{at}vanderbilt.edu1,2
- 1Division of Rheumatology, Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN 37232, USA
- 2Department of Microbiology and Immunology, Vanderbilt University School of Medicine, Nashville, TN 37232, USA
- 3Jyväskylä Central Hospital, 40620 Jyväskylä, Finland
- 4Division of Rheumatic Diseases, Department of Internal Medicine, University of Texas Southwestern Medical School, Dallas, TX 75390, USA
Abstract
In order to test the ability of peripheral blood gene expression profiles to predict future disease severity in patients with
early rheumatoid arthritis (RA), a group of 17 patients (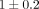 years disease duration) was evaluated at baseline for gene expression profiles. Disease status was evaluated after a mean
of 5 years using an index combining pain, global and recoded MHAQ scores. Unsupervised and supervised algorithms identified
“predictor genes” whose combined expression levels correlated with follow-up disease severity scores. Unsupervised clustering
algorithms separated patients into two branches. The only significant difference between these two groups was the disease
severity score; demographic variables and medication usage were not different. Supervised T-Test analysis identified 19 “predictor genes” of future disease severity. Results were validated in an independent cohort
of subjects of established RA with using Support Vector Machines and K-Nearest-Neighbor Classification. Our study demonstrates
that peripheral blood gene expression profiles may be a useful tool to predict future disease severity in patients with early
and established RA.
years disease duration) was evaluated at baseline for gene expression profiles. Disease status was evaluated after a mean
of 5 years using an index combining pain, global and recoded MHAQ scores. Unsupervised and supervised algorithms identified
“predictor genes” whose combined expression levels correlated with follow-up disease severity scores. Unsupervised clustering
algorithms separated patients into two branches. The only significant difference between these two groups was the disease
severity score; demographic variables and medication usage were not different. Supervised T-Test analysis identified 19 “predictor genes” of future disease severity. Results were validated in an independent cohort
of subjects of established RA with using Support Vector Machines and K-Nearest-Neighbor Classification. Our study demonstrates
that peripheral blood gene expression profiles may be a useful tool to predict future disease severity in patients with early
and established RA.
1. Introduction
Rheumatoid arthritis (RA) is a chronic, inflammatory joint disease with autoimmune features. Substantial evidence suggests that early intervention in individuals with RA results in improved control of disease activity, decreased joint damage, and fewer extraarticular manifestations [1–3]. Early RA patients may benefit from early aggressive therapies, such as new biologic agents that block the activity of TNF-α (Tumor Necrosis Factor alpha), which control disease activity and joint destruction [4, 5]. However, these biologic agents are generally expensive and up to 30% of RA patients have incomplete responses [6, 7]. These drugs also have significant side effects including increased severe infection and other autoimmune manifestations. In the approximately 30% of early RA patients who do not develop erosions [7], treatment with TNF blockers may not be necessary, and other drugs such as methotrexate may be sufficient. These clinical issues highlight the need for new approaches that would permit individualization of therapy for patients with early RA including development of additional prognostic markers.
Current prognostic methods for RA are generally based on the integrated use of information derived from patient self-assessment questionnaires, the physical examination, routine laboratory studies such as (erythrocyte sedimentation rate) ESR and (C-reactive protein) CRP and radiographic findings. Titer of rheumatoid factor and anti-CCP (antibodies to cyclic citrullinated peptides), imaging methods, including conventional X-rays, ultrasound and magnetic resonance, and genetic markers, such as HLA-DRB1 alleles, have also been employed [8–12]. Most diagnostic methods are dependent on the diagnostic tests that are evaluated. This may result in circularity and overestimation of the diagnostic properties of the tests [13–15]. Considering the prevalence of RA, which is estimated as 1% of the US population (NIAMS, National Institute of Arthritis and Musculoskeletal and Skin Diseases 2004), even a small increase in the accuracy of disease severity prediction has the potential to benefit a substantial number of RA patients.
Microarrays provide a powerful tool to screen expression levels of thousands of genes in single samples. We and others have used this approach to identify gene expression signatures in peripheral blood mononuclear cells (PBMCs) of individuals with autoimmune diseases, including RA, systemic lupus erythematosus [16], multiple sclerosis, and type I diabetes mellitus [17–21]. We also have described a unique gene expression signature that distinguishes patients with early RA from those with more established disease [22]. The objective of the present study was to determine if gene expression signatures collected early in the course of RA could predict future disease severity.
2. Materials and Methods
2.1. Patients
The 17 patients with early RA and 9 patients with established RA used for this study were included in previous reports [17–21]. We collected blood samples from 17 patients with early RA and 9 patients with established RA and analyzed differential
gene expression profiles using microarrays at an earlier date (2001–2002). At the time of blood collection, the mean SEM disease
duration of early RA patients was 1 ± 0.2 years and of established RA patients was 10 ± 2 years. Follow-up clinical information
was obtained by one of the investigators (T.S.) as part of ongoing longitudinal investigations. Clinical evaluations were
performed 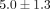 years after blood collection and microarray analysis. Self-assessment evaluations collected at the time of the follow-up
analysis included 100 mm visual analog scales for pain and global assessment and a modified health assessment questionnaire
(recoded MHAQ). Each of these measures was converted to an indexed score. “Pain” and “Global”: 0 = 0–9, 1 = 10–29, 2 = 30–59,
and 3 = ≥60; “Recoded MHAQ”: 0 = 0, 1 =
years after blood collection and microarray analysis. Self-assessment evaluations collected at the time of the follow-up
analysis included 100 mm visual analog scales for pain and global assessment and a modified health assessment questionnaire
(recoded MHAQ). Each of these measures was converted to an indexed score. “Pain” and “Global”: 0 = 0–9, 1 = 10–29, 2 = 30–59,
and 3 = ≥60; “Recoded MHAQ”: 0 = 0, 1 =  , 2 =
, 2 =  , 3 =
, 3 =  , and 4 = >1.0. An overall composite index score was then calculated as a sum of the three-score components. The overall disease
score had a possible range of 0–10, with scores of 0–3 considered mild and scores of 4–10 classified as severe. Determination
of clinical course was blinded to results of the microarray analysis.
, and 4 = >1.0. An overall composite index score was then calculated as a sum of the three-score components. The overall disease
score had a possible range of 0–10, with scores of 0–3 considered mild and scores of 4–10 classified as severe. Determination
of clinical course was blinded to results of the microarray analysis.
Vanderbilt University Institutional Review Board approved this study. All participants provided written informed consent.
2.2. Sample Preparation and Microarray Procedures
PBMC were isolated from 20 mL heparinized blood on a Ficoll-Hypaque gradient. All samples were processed within 2–4 hours of blood collection. Total RNA was isolated with Tri-Reagent (Molecular Research Center. Inc., Cincinnati OH) and 5 μg RNA was used to prepare cDNA with reverse transcriptase (Superscript II, Invitrogen Corporation, Carlsbad, CA) in the presence of 33P-dCTP. Labeled probes were purified using a Bio-Spin 6 Chromatography Column (Bio-Rad Laboratories, Inc., Hercules, CA). Before hybridization, GeneFilters membranes (GF-211, Research Genetics/Invitrogen Corporation, Carlsbad, CA) were washed in boiled 0.5% SDS, saturated with 5.0 mL Microhyb solution (HYB125.GF, Research Genetics/Invitrogen Corporation, Carlsbad, CA). Filters were treated with prehybridization reagents (5.0 μg Human Cot-1 DNA and 5.0 μg Poly dA, Invitrogen Corporation, Carlsbad, CA) in a hybridization roller tube (Midwest Scientific, St. Louis MO) for 2 hours at 42°C. Purified, labeled probes were denatured and added to roller bottles containing filters and prehybridization solution. GeneFilters membranes were hybridized overnight at 42°C. After hybridization, membranes were washed three times, exposed to imaging screens for 24 hours and screens were scanned by a phosphorimager (Molecular Dynamics/Amersham Biosciences, Piscataway NJ). Acquired images were loaded into Pathways 4.0 software (Research Genetics/Invitrogen Corporation, Carlsbad, CA). The relative intensity of each spot on the membrane was determined and the microarray dataset was subjected to further analysis using the different analytical platforms. Data were normalized to yield an average intensity of 1.0 for each clone (4133) represented on the microarray. Reproducibility of the method was established by performing replicate hybridizations to separate microarrays. Original microarray data are deposited in the GEO database, accession number GSE1964 (GSM35124-GSM35142).
2.3. Data Analysis
Cluster (version 3.0) and TIGR microarray software MultiExperiment Viewer (MEV) were used to identify genes whose expression levels differed significantly among the sample groups. The following data analysis modules of MEV were used to perform further analyses: HCL (hierarchical clustering), ST (support tree clustering), supervised T-Test, SVM (support vector machines), KNNC (K-Nearest-Neighbor Classification), and (principal components analysis) PCA. Detailed descriptions of the applications of these programs to the analysis are provided in the results section. Analysis procedures presented here comply with (minimal information about a microarray experiment) MIAME guidelines established by the Microarray Gene Expression Data Society (http://www.mged.org/). Clinical variables are shown as mean ± SEM. Statistical analyses of the clinical data were carried out using Fisher's exact test or Student's T-Test with a P value of <.05 considered significant.
3. Results
3.1. Clustering Analysis of ERA Patient Gene Expression Profiles
We performed unsupervised hierarchical clustering of gene expression profiles of ERA patients. First, the 4133 genes for which we had expression data were filtered at a standard deviation of 2 using Cluster software. A total of 192 genes passed this filtering condition. Unsupervised hierarchical clustering using this 192-gene expression profile segregated patient samples into two major groups (Figure 1). We compared patient clinical features to determine if the patients that segregated into the two clusters exhibited any common characteristic (Table 1). Age of disease onset, race, gender, and presence or absence of rheumatoid factor were not statistically different between individuals in the two groups. Medication uses of steroids and DMARDs or their combinations at the time of sample collection were also not statistically different. The only significant difference was the disease index distribution. In Cluster 1, 8 of the 10 patients (80%) developed mild disease (green bar) according to the clinical evaluation 5 ± 1.3 years after patient samples were collected, while in Cluster 2, only 1 of 7 (17%) had developed mild disease (P = .015). The others in Cluster 2 (6 of 7) were classified as having severe disease (red bar) at follow-up. Classification of disease severity was performed without knowledge of the results of the hierarchical clustering patterns identified by analysis of gene expression profiles. These results suggest that gene expression profiles can be used to predict future disease severity.
Clinical phenotypes corresponding to cluster designations.
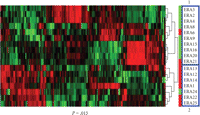
Unsupervised hierarchical clustering was applied to the expression profile of 192 genes using the filtering condition (SD = 2). Patients segregated into two clusters: 1 and 2. Difference of distribution of severe or mild patients in the two clusters was significant, P = .015, Fisher's Exact Test.
3.2. Supervised T-Test Identifies Predictor Genes
Next, we employed a supervised method of analysis to identify genes within the 4133-gene microarray database whose expression profiles predicted future disease severity to permit us to set certain criteria prior to the gene identification process. The supervised T-Test method was used to identify genes that were differentially expressed according to their disease severity (Figure 2). For this analysis, we divided patients into the severe disease group and the mild disease group according to their clinical evaluation (see Section 2.1) and set the following criteria for the genes: difference in expression of identified genes between the two groups had a P value <.001 with the multiple test correction applied (adjusted Bonferroni correction). Statistic t was calculated based upon 500 permutations for each gene across the 17-patient samples. Under these stringent conditions, 19 genes were identified.
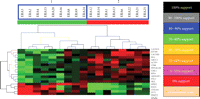
Support tree hierarchical clustering was applied to the expression profiles of 19-predictor genes among the 17 patients. A. Jackknife resampling was used with permutation 500 times. The color codes correspond to a given level of support. Branchs 1 and 2 are significantly separated according to their disease severity (P = .00004, Fisher's Exact Test).
We performed support tree clustering using expression data of these 19 genes with Jackknife algorithm resampling with 500 permutations. Support tree clustering not only identifies hierarchical trees but also calculates and shows the statistical reliability or support for the cluster of the trees, based upon the Jackknife resampling of the data. Jackknife resampling takes each gene expression profile across all the patients and randomly omits a patient. This method produces an expression profile that has all patients minus one, which minimizes the effects of single outlier values. For each resampling process, a hierarchical cluster is determined and compared to the original clustering result. The percentage of the original clustering results that occur during the number of resamplings indicates the level of reliability or support for the clustering result. Two major branches were produced from this analysis (Figure 2). All patients with future mild disease severity were in one branch and all patients with future severe disease severity were in the other branch. Therefore, the support tree results from Jackknife resampling indicate that there was no obvious influence of outliers on gene identification and the clustering profile.
3.3. The 19-Predictor Genes
The 19-predictor genes could be divided into two groups depending upon whether they exhibited higher or lower expression in the future mild and future severe disease groups, respectively (Figure 3). Normalized polished expression data of these 19 genes in the severe and mild groups (Figure 3(a)) and original expression data without normalization (Figure 3(b)) reveals similar expression patterns in the future severe and mild groups. Among the 19-predictor genes: FVT1 (follicular lymphoma variant translocation 1), EHD1 (EH-domain containing 1), COL4A1 (collagen, type IV, alpha 1), PRMT2 (protein arginine methyltransferase 2), and TFCP2 (transcription factor CP2) were underexpressed in the severe patient group compared to the mild patient group, the other genes: FHL3 (four and a half LIM domains 3), SKIL (SKI-like oncogene), RPIA (ribose 5-phosphate isomerase A (ribose 5-phosphate epimerase)), SPRY2 (sprouty homolog 2 (Drosophila)), F2RL1 (coagulation factor II (thrombin) receptor-like 1), PPP1R12B (protein phosphatase 1, regulatory (inhibitor) subunit 12B), LTBR (lymphotoxin beta receptor (TNFR superfamily, member 3)), GADD45A (growth arrest and DNA-damage-inducible, alpha), ARHGEF16 (Rho guanine exchange factor (GEF) 16), MLL (myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila)), ACYP1 (acylphosphatase 1, erythrocyte (common) type)), EIF3S9 (eukaryotic translation initiation factor 3, subunit 9 eta, 116 kDa), CACNB2 (calcium channel, voltage-dependent, beta 2 subunit), and ABCC3 (ATP-binding cassette, subfamily C (CFTR/MRP), member 3) were overexpressed in the future severe patient group compared to the future mild patient group.
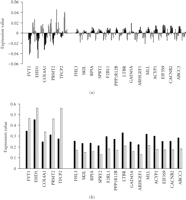
Identity and expression levels of the 19 genes in individuals comprising the future mild and future severe disease groups are shown by bar graphs. (a) The normalized polished expression data of these 19 genes in severe and mild groups. (b) The original expression data without normalization in the future severe and mild groups for the 19-predictor genes. Note that standard deviations are not shown because the t statistic was calculated based on permutations. Black bars represent the severe group, white bars represent the mild group.
3.4. Independent Validation of Disease Severity Signature
To further explore properties of this 19-gene expression profile, we applied microarray profiling and clinical follow-up to an independent cohort of patients with established RA (N = 9, average disease duration of 10 years at the time of expression profiling, 15 years at the time of clinical follow-up). Expression profiling of these RA patients was performed within the same time frame as the ERA patients. After normalization, expression data for the 19 genes identified above were extracted and analyzed using the hierarchical clustering algorithm (Figure 4(a)). This segregated the 9 RA patient samples into two clusters with 100% support and these two clusters exactly corresponded to their future clinical evaluation; group 1: severe disease, group 2: mild disease. Average expression values of these 19 genes were also determined for both the severe and mild groups. The expression differences for these 19 genes were identical to the expression pattern observed in ERA patients when segregated based upon disease severity (Figure 4(b)). FVT1, EHD1, COL4A1, and TPCP2 exhibited lower expression levels in the severe group of patients than in the mild group of patients. Conversely, FHL3, SKIL, RPIA, SPRY2, F2RL1, PPP1R12B, LTBR, GAD45A, ARHGEF1, MLL, ACYP1, EIF3S9, CACNB2, and ABCC3 exhibited higher expression levels in the severe group of patients compared to the mild group of patients. This mirrors what was found in the ERA group (Figure 3(b)).
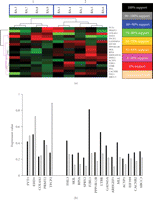
Hierarchical classification of an independent established RA cohort using the 19-gene expression profile. (a) Support tree hierarchical clustering was applied to the expression profile of the 19-predictor genes in 9 RA patients. Jackknife resampling was used with 500 permutations. The color codes correspond to a given level of support. Branch 1 and 2 are significantly separated according to their disease severity. (P = .008, Fisher's Exact Test). (b) The original expression data without normalization in the severe and mild groups for the 19-predictor genes in 9 RA patients. Note that standard deviations are not shown because the t statistic was calculated based upon permutations. Black bars represent the severe group and white bars represent the mild group.
We employed PCA to examine the ability of the 19-gene expression profile to discriminate between the combined RA cohorts,
ERA and established RA, based upon future disease severity. PCA projected the patients into a two-dimensional plane according
to their 19-gene expression profile (Figure 5). In the two-dimensional plane (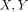 ), patients segregated into two areas that are separated by the dashed line. The distribution of the patients in 2D space
determined by their 19-gene expression profile by PCA analysis indicates that the 19-gene predictor system can segregate the
two independent RA patient groups solely upon their future disease severity rather than other parameters such as disease duration.
), patients segregated into two areas that are separated by the dashed line. The distribution of the patients in 2D space
determined by their 19-gene expression profile by PCA analysis indicates that the 19-gene predictor system can segregate the
two independent RA patient groups solely upon their future disease severity rather than other parameters such as disease duration.
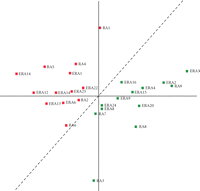
PCA classification of ERA and established RA patients by the 19-gene predictor. Two-dimensional projection of 26 patients
based upon their gene expression levels of 19-predictor genes onto the (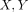 ) plane. Green squares represent the future mild disease group and red squares represent the future severe disease group.
The dashed line segregates the future severe and mild groups of RA patients with 100% accuracy.
) plane. Green squares represent the future mild disease group and red squares represent the future severe disease group.
The dashed line segregates the future severe and mild groups of RA patients with 100% accuracy.
We employed SVM and KNNC methods to determine if disease severity in the established RA patients could be predicted based upon expression profiles of the 19-gene expression set identified from analysis of the ERA patient set. ERA patients were used as the training set and each ERA patient was assigned to the severe or mild group according to both their corresponding 19-gene expression signature and their clinical evaluation. After SVM training, the SVM algorithm was applied to classify the 9 RA patients using their 19-gene expression value. The 9 RA patients were accurately segregated into the two groups according to their clinical evaluation with one exception, RA no. 1. RA no. 1 should be in the severe group according to clinical evaluation, but SVM classified it as mild status according to the gene expression profile (Table 2). Similarly, KNNC was applied to classify the independent 9 RA patients. They were separated into two groups by KNNC exactly according to their clinical evaluation (Table 3). Therefore, with this independent data set, we confirmed that expression profiles of these 19 genes represent a disease severity signature in RA.
SVM analysis accurately predicts future disease severity in RA patients using ERA 19-gene expression profiles as the training set.
KNNC analysis accurately predicts future disease severity in RA patients using ERA 19-gene expression profiles as the training set.
4. Discussion
Disease-modifying therapy early in the course of RA can lead to improved disease control and decreased joint damage. However, the uncertain course of RA in some patients coupled with the adverse effects and high cost of newer therapies make decisions regarding treatment strategies complex. About 30% of early RA patients will not develop severe disease and a small number may even undergo remission without treatment. The current prognostic factors are relatively powerful tools, including measurement of health assessment questionnaire (HAQ) scores, autoantibody levels, and genetic markers. Although the prognostic sensitivity of the combined tests is in range of 80%–90%, this may not be sufficient to predict outcome in an individual patient [7, 25–27]. Gene expression profiling strategies have been widely used in cancer studies for purposes of diseases classification, evaluation of responses to therapies, and prediction of disease outcome [23, 24, 26, 28–39]. Similar gene expression profiling strategies have been employed in autoimmune disease, including RA, to address similar questions [17, 19–22, 28, 29]. For example, a set of indicator genes has been identified that predict responses to the TNF-α blocking agent, infliximab, in RA [30]. Supervised algorithms were applied to identify responder genes, whose expression levels discriminate between those subjects who would respond to infliximab therapy and those subjects who were poorly responsive to infliximab therapy. Several genes have also been identified whose expression levels correlate with current disease activity based upon standard measurements of disease activity including the HAQ score, CRP levels, ESR, and rheumatoid factor [31] levels [29]. A general view is that gene expression levels may provide a more quantitative index of disease activity than currently available. Although studies such as these must be validated in larger patient cohorts, these results suggest it may be possible to employ differences in gene expression to estimate disease activity.
Here, we wanted to determine if gene expression profiling is a method also able to predict future disease severity in RA. To explore this question, we compared the clinical status of subjects with RA to expression data obtained from these subjects early in their disease. We applied the supervised T-Test algorithm to identify a combination of 19 genes whose past expression levels predicted their future clinical course in a group of 17 patients with early RA. We achieved 100% (17/17) accuracy of prediction of future disease severity if our clinical follow-up assessment is 100% accurate.
An independent dataset is optimal to further validate 19-gene prediction system. In this regard, we tested the 19-gene predictor system using an independent RA data set. We applied two supervised methods: SVM and KNNC to validate our results. SVM [32, 33, 40–42] and KNNC [28, 34–37] are supervised machine learning algorithms used in gene expression profiling studies. SVM uses kernel function to build classification rules and KNNC uses weighted voting to designate the class of test samples. There is no general consensus for which method is superior. Therefore, we applied both methods to our analysis and both methods produced almost 100% accuracy for prediction of future disease severity in RA patients. Our results from SVM and KNNC are consistent with our clustering analysis and PCA grouping. Thus, by using an independent cohort of patients with established RA, we were able to further confirm that the expression profile of these 19-predictor genes represents a signature of disease severity in RA. These results also support the notion that the expression pattern of these 19 genes is stable as a function of disease duration. The expression pattern exists in both early and established RA patients. Therefore, this test may have application at any point during the disease history of an individual with RA. In the independent validation, only one RA subject, RA no. 1, was not consistently classified according to the disease severity status. However, this patient may develop severe disease later and bears close monitoring.
Of the 19 genes comprising our predictor system, five are underexpressed in patients who develop a severe disease phenotype and fourteen genes are overexpressed. At present, we cannot conclude if expression levels of these genes are causal factors contributing to the differential disease severity or are indicators of future disease activity since the mechanistic basis of future disease activity is not well understood. It is tempting to speculate that better understanding the impact of alterations in expression levels of this group of genes may not only improve our ability to predict future disease activity in RA but may also contribute to our understanding of mechanisms leading to more severe disease. For example, SKIL (SKI-like oncogene), overexpressed in the severe disease group, is induced by TGF-β1, whose expression level is positively correlated with serum levels of CRP, a clinical marker of disease activity and severity [38]. Further, increased expression of F2RL1I (also named PAR2, protease-activated receptor 2) in a murine model of arthritis correlates with joint swelling and erosion. Inhibition of increased expression of PAR2 substantially reduces inflammatory responses in the joint [39]. Expression levels of PAR2 are also substantially increased in RA synovium compared to control synovial tissue. Spontaneous release of inflammatory cytokines is substantially inhibited by a PAR2 antagonist in a dose-dependent manner [39]. These results suggest that increased expression of F2RL1 (PAR2) may contribute to increased disease activity in RA patients. Although the mechanistic significance of how differences in expression levels of these 19-predictor genes may influence future disease severity in RA is not entirely clear, our results clearly demonstrate that these differences in expression may have utility in predicting future disease outcome.
At the time of sampling, between sampling and clinical follow-up, and at the time of clinical follow-up, all patients in our study were on some type of antirheumatic therapy. Clinically, RA is a very heterogeneous disease and our system may be able to discriminate between individuals who will develop aggressive or mild disease. An alternative interpretation is that expression levels of these 19 genes actually discriminates between individuals who exhibit good responses to antirheumatic therapies and therefore develop mild disease and those patients who exhibit poor responses to antirheumatic therapies and therefore develop a severe disease course. Further studies are needed to determine if expression levels of these 19-predictor genes actually forecast poor responsiveness to therapy rather than aggressive versus mild disease.
In this analysis, we considered association of the RA covariates, disease severity, race, gender, Rf titer, and medication usage and found strong association of the 19-predictor gene signature with disease severity. Although all subjects were on medications, they did not receive any specific treatment at the exact time of their blood draw. Other RA covariates, such as the HLA-DRB1 shared epitope [10], age of disease onset, anti-CCP titer, education status, tobacco usage, were not considered in our analysis. These are important covariates to be considered in future studies.
One advantage of our 19-gene predictor system is that we used PBMC as a resource for gene expression profiling even though PBMC are not localized to the site directly affected by the disease of RA. PBMC are a very easily accessible human tissue sample compared to a surgical biopsy of an affected tissue. Therefore, PBMC may be a good common resource for disease related “biomarkers” identification. PBMC have been widely used as a resource for gene expression profiling in RA [17, 21, 22, 28, 30]. PBMC also may represent a suitable common source for gene expression profiling experiments in other diseases such as certain cancers or chronic noninflammatory diseases whose major affected sites are not blood.
In our analysis, we identified and selected expression levels of 19 genes as predictors of future disease severity. Other investigations using gene expression profiling methods for disease prediction or classification have used more genes in their classifying system [20, 28, 37]. Expression levels of a greater numbers of genes in a classification system have the potential to produce more accurate results. However, our 19-gene predictor system achieves 100% accuracy in an independent test dataset. This indicates that our 19-gene system is reliable. Further, fewer genes in a classification system can facilitate future clinical implementation and save potential costs for manufacturing such a test and ease economical burdens. In summary, our 19-gene predictor system accurately discriminates between subjects with RA who will develop severe disease and those who will develop mild disease with 100% accuracy. These results suggest that it is possible to predict future disease severity using this type of approach. We propose that addition of this analysis to other measures, such as HAQ score, HLA genotyping, and rheumatoid factor may be useful to predict future disease severity in early RA patients. This information may be useful for designing tailor-made therapies for RA patients.
Acknowledgments
This work was supported by Grants: AI053984 and AI044924 from the NIH and the McGee Foundation. The first author is currently at The University of Texas, Austin, and the third author is currently at Stanford University School of Medicine. This work was funded in part by a Grant from the NIH to ArthroChip.
- Received July 27, 2008.
- Revision received December 16, 2008.
- Accepted March 11, 2009.
- © 2009 Zheng Liu et al.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
-
- Morel J., j-morel{at}chu-montpellier.fr
- Roch-Bras F.,
- Molinari N.,
- Sany J.,
- Eliaou J. F.,
- Combe B.
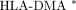 0103 and
0103 and 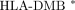 0104 alleles as novel prognostic factors in rheumatoid arthritis”. Annals of the Rheumatic Diseases, vol. 63 no. 12 pp. 1581–1586 2004 doi:10.1136/ard.2003.012294.
0104 alleles as novel prognostic factors in rheumatoid arthritis”. Annals of the Rheumatic Diseases, vol. 63 no. 12 pp. 1581–1586 2004 doi:10.1136/ard.2003.012294. - ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵















