Transcriptional Mechanisms Underlying Addiction-Related Structural Plasticity
Abstract
Drug addiction is marked by long-lasting changes in behavior that result, in part, from altered patterns of gene expression within limbic forebrain regions, such as the nucleus accumbens (NAc). These changes in gene transcription are coordinated by a complex series of histone modifications surrounding DNA that result in either repression or activation of gene expression. Recent evidence has identified a network of gene expression changes, regulated by the transcription factor ΔFosB, which alter the structure and function of NAc medium spiny neurons to control addictive-like behavior. In this review, we will discuss recent advances in our understanding of chromatin regulation by cocaine, as well as the consequences of such regulation on structural plasticity and its functional relevance to drug addiction.
Introduction
Chronic exposure to drugs of abuse results in altered patterns of gene expression throughout the reward circuitry of the central nervous system (CNS); however, the time course of such events does not fully explain the long-lasting behavioral syndromes associated with drug addiction (1–3). Given this finding, many have argued that the initial biochemical and transcriptional changes induced by chronic drug exposure may ultimately result in longer-lived alterations in neuronal structure and morphology in diverse cell types throughout the limbic forebrain (4, 5). Such neuronal restructuring, which can persist from months to years following cessation of drug administration (4, 6), has been proposed to underlie the persistent behavioral phenotypes associated with addiction. Although the mechanisms responsible for such long-lasting changes in neural circuitry remain largely unknown, recent studies have identified a transcriptional mechanism through which posttranslational modifications of histones at the fosb promoter increase ΔFosB, a highly stable splice product of the immediate early gene fosb, thus regulating its downstream transcriptional targets, including nuclear factor–kappa B (NF-κB), cyclin dependent kinase 5 (Cdk5), and myocyte enhancing factor 2 (MEF2) to enhance various forms of structural plasticity (7–10). In this review, we will discuss this novel transcriptional process in the context of structural plasticity and its relevance to the promulgation of associated addiction-related behaviors.
Addictive Drugs Regulate Synaptic and Structural Plasticity
Almost all classes of addictive substances alter the structure and connectivity of diverse neuronal populations throughout the brain (4, 5). Among the most heavily characterized class of abused drugs, psychostimulants can increase the dendritic branching and/or dendritic spine number on neurons in the medial prefrontal and orbitofrontal cortices, in the striatum [including both the caudate putamen (CPU) and the nucleus accumbens (NAc)], and in dop-aminergic projection neurons originating from the ventral tegmental area (VTA). Given the degree and potential stability of restructuring that occurs throughout the brain following chronic drug administration, many have argued that these adaptations may be responsible for long-term behavioral syndromes, which can ultimately result in relapse-related behaviors following periods of drug abstinence. Furthermore, several studies of structural plasticity in animals self-administering drugs of abuse (SA) versus animals receiving experimenter-administered drugs (EA) demonstrate that certain changes in spine structure require volitional control over drug intake (Figure 1). Moreover, as a general rule, structural plasticity is greater in SA than in EA animals, which coincides with differences in activation of neurocircuits associated with motivation and reward (11). Thus, this differential regulation of plasticity may enable selective targeting of motivation-related pathways to control addiction liability (4, 5).

Structural plasticity of major cell types in the neural circuitry underlying addiction. Ventral tegmental area (VTA) dopamine projection neurons (solid purple lines) impinge directly upon nucleus accumbens (NAc) and medial prefrontal cortex (mPFC) neurons, as well as on amygdala and hippocampal neurons (the latter projections are not shown in the figure). GABAergic afferents (some direct, some indirect) from the NAc to the VTA (solid blue lines) provide feedback to VTA dopamine neurons. Glutamatergic afferents to the NAc from mPFC, amygdala, and hippocampus (dotted blue lines) release glutamate directly onto medium spiny neuron spine synapses. Each structure contains specialized neuronal cell types that play a critical role in the regulation of addiction-like behavior. The major cell types, color-coded in the key, include amygdala (green) and NAc (blue) spiny neurons, PFC (orange) and hippocampal CA3 (red) pyramidal neurons, and VTA dopamine neurons (purple). Drugs of abuse can alter the structure of each of these cell types as defined in the table above. oPFC, orbital prefrontal cortex. Modified from (5, 85) with permission.
These forms of structural plasticity result from reorganization of the actin cytoskeleton at excitatory synapses and reflect the foundation for other synaptic changes, which, not surprisingly, coincide with functional changes in the electrophysiological properties of NAc medium spiny neurons (MSNs). These changes are highly time-dependent and likely contribute to long-term addiction-related behavioral plasticity. For example, within twenty-four hours following the last exposure to a chronic regimen of cocaine, there is an increase in thin (more highly plastic) spines, as well as an increased state of synaptic depression (12, 13). This increase in thin spines may represent a larger pool of silent synapses in this brain region (14, 15). Silent synapses contain N-methyl D-aspartate receptors (NMDARs) [but few-to-no α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptors (AMPARs)]; express relatively stable NMDA receptor-mediated excitatory postsynaptic currents (EPSCs); and are ideal substrates for long-term potentiation (LTP) (15, 16). Following cocaine treatment, such silent synapses in the NAc appear to express an increased proportion of NR2B-containing NMDA receptors (14), a finding consistent with these synapses being fairly immature (17, 18). During the course of cocaine withdrawal, these recently formed spines appear to be highly transient and may retract or consolidate into mushroom-shaped spines (12), events that are accompanied by an increase in the surface expression of GluR2-lacking AMPARs and a potentiation of these glutamatergic synapses (19–21). Behaviorally, during withdrawal from cocaine self-administration, there is evidence for an incubation of cocaine craving, characterized by a gradual and progressive increase in cocaine seeking that may require such changes in the stoichiometry of synaptic AMPARs (21). Additionally, recent evidence has indicated that re-exposure to cocaine following extended withdrawal results in a reduction in spine head diameter (12), a decrease in the surface expression of AMPARs (19), and a subsequent depression of strength at these synapses (22). Taken together, these data point to a complex interaction between spine-head structure, electrophysiological properties of NAc MSNs, and addiction-related behavior. Given that many synaptic proteins can regulate these events, it is important that we identify the precise transcriptional mechanisms involved in their regulation and how these changes guide behavior.
Chromatin Regulation and Gene Transcription
At its most basic level, chromatin functions both spatially and temporally to ensure the proper storage, organization, and output of genetic information during cellular differentiation and organismal development (23). Shown in Figure 2 is the nucleosome core particle, which represents the fundamental repeating transcriptional unit of chromatin, composed of 147 base-pairs of DNA wrapped around a core octamer of histone proteins (two copies each of H2A, H2B, H3, and H4, or variants of these proteins) (24). Nucleosomal structural variations that may be introduced via a number of tightly regulated processes, including covalent post-translational modifications of histones, chromatin remodeling complexes, and histone variant deposition, promote alterations in chromatin compaction that correlate with more “open” (euchromatic) versus “closed” (heterochromatic) transcriptional states; such chromatin regulation often results, respectively, in more “active” vs “inactive” states of gene expression (25–27). For example, histone modifications that weaken the interaction between histones and DNA, such as histone acetylation, act to neutralize positive charges on ɛ-amino groups of lysine residues on N-terminal histone tails and have long been correlated with transcriptional activation (25, 28). Likewise, histone deacetylation, which often strengthens the interaction between histones and DNA, promotes a state of transcriptional repression (29). Combinations of acetylation and numerous other post-translational modifications of histones, including phosphorylation, methylation, sumoylation, etc., have been shown to alter chromatin condensation, leading to altered levels of gene expression in cells (27, 30). These events occur through a dynamic and complex system of enzymatic processes that involve “writer” proteins (i.e., enzymes that catalyze the addition or removal of histone marks at specific substrates) and “effector” proteins with distinct “reader” modules (i.e., regulatory proteins that contain unique domains capable of recognizing specific histone modifications or combinations thereof) (27, 31–33). Unlike acetylation, histone methylation is unique in terms of its relative stability and complexity of regulation (34). As with the addition of acetyl groups to N-terminal histone tails, methyl groups can also be added to the ɛ-amino groups of lysine and arginines; however, methylation does not alter the charge of target residues and can exist in a multivalent state (i.e., histone lysine and arginine residues can be mono-, di- or tri-methylated). Furthermore, both the specific amino-acid residues being methylated, as well as the valency of methylation at those residues, will ultimately determine whether a particular methylation event is transcriptionally activating or repressive at a gene promoter (34). Numerous enzymes have been identified, which specifically methylate or demethylate particular lysine or arginine residues, as well as control the valency of methylation at those residues, thereby introducing a layer of incredible enzymatic complexity to this process of transcriptional regulation (34–36). Because histone methylation does not affect electrostatic interactions between histone proteins and DNA, the consequence of methylation (i.e., activating vs repressing) is largely determined by effector proteins that contain distinct binding motifs, such as “plant homeodomain” (PHD) fingers and chromodomains, that recognize specific methyl marks with remarkable precision to promote various downstream transcriptional events (33). By gaining a better understanding of these chemical modifications on histone proteins, we are uncovering the role of transcriptional plasticity in the brain during the development of complex behavioral and pathological states, such as drug addiction. This provides hope that future classes of therapeutic agents aimed at targeting these mechanisms may be more effective in treating addiction.
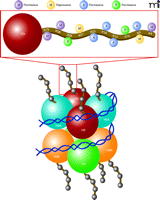
Chromatin modifications on histones regulate gene expression patterns. Chromatin functions to ensure the proper storage, organization and output of genetic information. Shown is the nucleosome core particle, which represents the fundamental repeating transcriptional unit of chromatin, composed of 147 base pairs of DNA wrapped around a core octamer of histone proteins (two copies each of H2A, H2B, H3, and H4, or variants of these proteins). Nucleosomal structural variations are regulated by covalent post-translational modifications of histones, leading to alterations in chromatin compaction leading to more “open” (euchromatic) versus “closed” (heterochromatic) transcriptional states. Combinations of acetylation and numerous other post-translational modifications occurring on N-terminal histone tails (including phosphorylation, methylation, sumoylation, etc.) have been shown to alter chromatin condensation leading to altered levels of gene expression in cells. As shown above, histone modifications that weaken the interaction between histones and DNA, such as histone acetylation at K23, K18, K14, and K9, methylation at K79, K36, and K4, or phosphorylation at S28 and S10, are correlated with transcriptional activation. Likewise, histone deacetylation or histone methylation, which strengthens the electrostatic interaction between histones and DNA, of K27 or K9, promotes a state of transcriptional repression.
Chromatin Regulation of Synaptic Plasticity
Many of the enzymes controlling chromatin dynamics participate in transcriptional networks to control synaptic plasticity in both the developing and adult CNS (37–39). For example, loss of lysine (K)-specific demethylase 5C (SMCX) activity, which regulates histone 3 lysine 4 (H3K4) tri-demethylation, has recently been linked to mental retardation and epilepsy (40). These processes likely occur through disruption of SMCX’s repressive enzymatic activity, as well as by modulation of potential “reading” capabilities contained within its two PHD finger domains, which may act to stabilize SMCX in chromatin remodeling complexes (40–44). SMCX also appears necessary for normal neuronal dendritic development, suggesting a direct link between chromatin function and synaptic development, maintenance, or both (40). Similarly, mutations that disrupt the interaction between methyl CpG binding protein 2 (MeCP2), an essential regulator of DNA methylation, which has been causally linked to Rett syndrome, and the DNA helicase/ ATPase/histone variant chaperone alpha thalassemia/mental retardation X-linked (ATRX), promote pathologies associated with these epigenetic regulators, leading to abnormalities in dendritic structural morphology (45). These studies suggest that both the failure to remove or add epigenetic marks, as well as the inability to appropriately coordinate interactions between chromatin regulators, impair downstream neuronal gene expression, ultimately disrupting synaptic function and promoting the development of disease states. Other chromatin modifiers, including histone deacetylases 1 and 2 (HDAC1 and HDAC2) have been implicated in synaptic activity both throughout development and in adult animals. Developmental disruption of both HDAC1 and HDAC2 was demonstrated to facilitate excitatory synapse maturation and synapse number (46). Of note, deletion of HDAC2 in mature neurons appears sufficient to enhance basal excitatory neurotransmission, suggesting a lack of redundancy in the role of associated chromatin regulators in mediating various aspects of synaptic activity. Furthermore, neuronal specific HDAC2 overexpression in adult animals decreases synapse number, synaptic plasticity, and memory formation (47). These data are in accordance with findings that demonstrate a role for histone acetylation in the induction of long-term potentiation (LTP), whereby increased histone acetylation via HDAC inhibition results in enhanced LTP in mammalian neurons (48). Additionally, pharmacological manipulations known to alter long-term facilitation or long-term depression (LTD) are sufficient to produce alterations in histone acetylation at specific gene promoters in the nervous system. Such phenomena were first observed in Aplysia cell culture systems, where the administration of serotonin (5-HT), which enhances long-term facilitation, was demonstrated to promote CREB binding protein (CBP) recruitment to the CAAT box enhancer binding protein (C/EBP) promoter, thereby increasing histone acetylation and transcription factor binding upstream of the C/EBP transcriptional start site (TSS) (49).
Consistent with a role for chromatin regulation in the development of synaptic plasticity, as well as the observed correlations between transcriptional activation and behavioral plasticity, numerous reports suggests an important regulatory role for chromatin modifications in the development of behavioral memory. Because of the potential stability of certain histone/DNA modifications, drug-mediated alterations in chromatin regulation and synaptic plasticity might help to explain long-lasting alterations in the brain’s neural reward circuitries, and associated memory centers, which play an important role in the development of addiction-associated behaviors following repeated drug administration (23). Although very little is known concerning the role of chromatin regulation in the hippocampus following drug exposure, numerous learning and memory-related studies suggest important roles for histone acetylation in the development of long-term memory formation, whereby histone marks that confer transcriptionally active states appear to be essential to the development of such memories (39, 50). For example, mice deficient in the histone acetyltransferase (HAT) CBP, which catalyzes the addition of acetyl groups to histones, display memory deficits that can be reversed with administration of HDAC inhibitors (HDACis) (48, 51, 52). CBP also interacts directly with cAMP response element binding (CREB) to promote transcriptional activation of target genes, many of which have been demonstrated to directly enhance various forms of synaptic plasticity (53–55). Furthermore, induction of extracellular signal–regulated kinase (ERK) signaling as a direct consequence of contextual fear conditioning—which is believed to promote memory formation and synaptic plasticity—increases histone phosphorylation in the hippocampus, thereby enhancing transcriptional activation in this brain region (39, 50). More recently, it was demonstrated that disruption of experience-dependent acetylation on histone H4 (H4K12) in the hippocampus results as a consequence of normal cognitive aging, and correlates with age-dependent deficits in memory and memory-associated transcription (56). Transcriptional repression in the hippocampus is also necessary for memory formation and cognition, as demonstrated through changes in gene-specific DNA methylation patterns (57, 58), and altered levels of repressive histone methylation (59, 60). Generally, it appears that increasing transcriptional plasticity, through enzymatic chromatin activation, is necessary for the enhancement of behavioral memory, although such plasticity is likely occurring only on a subset of specific genes.
Chromatin Regulation of Addiction-related Plasticity
Altered patterns of gene expression throughout the reward circuitry of the brain following chronic drug exposure represent a key molecular mechanism believed to underlie both the transition from recreational drug use to the chronically “addicted” state and the persistence of addictive behaviors following cessation of drug intake (2, 61). One important site for such changes, as mentioned above, is the NAc, where alterations in drug-induced gene expression have been reported to persist following months of drug abstinence in rodent models, consistent with long-lasting behavioral abnormalities observed in animals after extended periods of drug withdrawal (62). It is important to note, however, that although such sustained changes in drug-induced gene expression have been reported, most drug-mediated transcriptional alterations appear to be rather transient, often temporally associated with instances of acute drug treatment or subsequent re-exposure following chronic administration of drugs. For example, recent data employing microarray analyses to investigate patterns of gene expression directly following drug exposure (i.e., one hour post-exposure) have revealed that although numerous genes display heightened levels of transcription immediately following acute administration of cocaine (i.e., a single dose), many more genes seem to be induced in the NAc of animals repeatedly exposed to the drug (7). Furthermore, not only do more genes display increased transcription following acute versus repeated cocaine treatment, but many of these same genes, were upregulated to a greater extent after repeated drug exposure. Such enhanced patterns of gene expression after repeated cocaine treatment was also observed after a period of withdrawal (i.e., seven days) followed by an acute drug re-exposure (7). Investigation of gene expression patterns immediately following drug administration, indicate that many of the behavioral differences observed between acutely and chronically treated animals (e.g., locomotor sensitization) are likely the result of enhanced levels of gene expression in the NAc. Because many of the genes known to be involved in regulating aspects of structural plasticity following repeated cocaine treatment (e.g., Cdk5, ΔFosB, etc.) are transcribed following chronic, but not acute, cocaine administration (63, 64), it is possible that “sensitized” transcriptional responses observed in chronically treated animals may result in more persistent neuroadaptations, such as alterations in NAc synapse number or strength, that ultimately affect behavioral responses to cocaine. Therefore, although a large portion of drug-induced gene expression changes do not persist for the length of the behavioral syndromes associated with drug addiction, many genes do become more sensitive to future transcriptional activation following a drug challenge after a period of withdrawal (7). As a result of these observations, it is clear that underlying molecular mechanisms must exist to promote stable changes in gene activation, transcriptional inducibility, and ultimately, long-lasting changes in neuronal structure and behavior. Indeed, chromatin-based mechanisms have been causally linked both to changes in gene expression and alterations in synaptic function that may underlie aspects of compulsive drug use.
Repeated exposure to all drugs of abuse is known to dramatically alter gene expression patterns throughout the reward circuitry of the brain. Chronic exposure to the psychostimulant cocaine is sufficient to induce alterations of histone acetylation, phosphorylation and methylation in the NAc. Thus, it is hypothesized that such modifications may be intricately involved in mediating behavioral sensitivity to drugs of abuse (61). The first in vivo evidence for this came from studies showing that genetic manipulation of specific HDACs, or pharmacological HDAC inhibition, dramatically affects animals’ behavioral responses to cocaine (65). For example, either systemic administration of two non-specific HDACis (sodium butyrate or trichostatin A) or intra-NAc delivery of the more-specific HDACi, suberoylanilide hydroxamic acid (SAHA), significantly potentiated the effects of cocaine as measured via conditioned place preference (CPP) experiments, reflecting an indirect measure of drug reward (65, 66). Similar potentiating effects of HDACis on drug-related behaviors have been observed with both dopamine D1-specific agonists and amphetamine (67, 68). It is noteworthy that HDAC inhibition also potentiates extinction from CPP and reduces CPP reinstatement, suggesting that HDAC inhibition, and thus potentiation of acetylation, may simply function to enhance behavioral saliency to a wide variety of environmental stimuli that require continual adaptation (69). Although, behaviorally, these data are consistent with a role for HDAC inhibition in the development of cocaine-induced neuroplasticity, data linking HDAC activity to alterations in cocaine-mediated structural or synaptic plasticity do not currently exist. Further complicating these studies is the fact that systemic administration of HDACis likely affects numerous cellular processes, which may complicate behavioral interpretations following drug exposure. For example, CPP is a relatively complicated behavioral task, in that it requires animals to associate contextual experiences with specific conditioned stimuli (e.g., cocaine), and therefore requires the activation and integration of numerous neural circuits (e.g., hippocampus, NAc, PFC, etc.) to promote behavioral outputs. When injected systemically, HDACis likely affect transcription in multiple limbic nuclei, making it difficult to ascertain whether observed behavioral outcomes are the result of universal alterations in learning and memory, or are specific to changes occurring in brain regions controlling motivated behaviors and reward (e.g., NAc). On the other hand, HDAC inhibition in the NAc is sufficient to enhance cocaine preference, indicating that the aforementioned behavioral effects elicited by HDACis are not the result of global deficits/enhancements in learning and memory.
Consistent with these data suggesting that increased histone acetylation results in increased drug sensitivity, CBP knockout mice exhibit reduced acetylation at the fosb promoter and reduced behavioral sensitivity to cocaine (70). Furthermore, viral overexpression of specific HDACs in the NAc (i.e., HDAC4 and 5, but not HDAC9) similarly decreased CPP—an effect dependent on the HDAC catalytic domain—whereas global HDAC5 knockout in HDAC5 null mice enhanced the rewarding effects of cocaine, a phenomenon that could be fully rescued by NAc-specific viral overexpression of the enzyme (66). The finding that certain HDACs, including HDAC5, may specifically control aspects of drug-mediated behavior has led to further investigations aimed at better understanding the mechanisms through which cocaine may exert its effects on histone acetylation patterns following drug exposure. Chronic, but not acute, exposure to cocaine induced HDAC5 phosphorylation in the NAc, leading to nuclear export, hyperacetylation, and increased expression of HDAC5 target genes, an event that has also been observed in animals chronically self-administering cocaine (66, 71). In addition, global genetic knockout of HDAC5 only results in increased cocaine preference in animals previously exposed to a sensitizing regimen of the drug, an effect that can be rescued by overexpression of HDAC5 specifically in the NAc. These data indicate that as animals are exposed to increasing levels of cocaine, HDAC5, which under “normal” conditions presumably acts to prevent aberrant environmentally-induced alterations in gene expression, becomes phosphorylated and is exported from the nucleus to promote the induction of numerous HDAC5 dependent transcripts that act to ultimately enhance behavioral responses to cocaine.
Although these alterations in HDAC phosphorylation and nuclear export occur in both EA and SA animals, systemic HDAC inhibition can result in different behavioral responses depending on the self-administration protocol being used. Such behaviors depend on the time in which HDACis are administered during the course of self-administration training. For example, rats treated with the HDACi Trichostatin A, prior to each fixed ratio 1 (FR1) scheduled self-administration session, lever-pressed for fewer infusions of cocaine during training sessions, which was interpreted to represent a decrease in the reinforcing value of the drug (72). On the other hand, animals that were trained to self-administer cocaine before receiving systemic administration of the HDACi NaBut displayed increased cocaine lever pressing, indicating that such treatments increase the reinforcing properties of the drug. The latter results are consistent with CPP and locomotor sensitization experiments suggesting that HDAC inhibition enhances behavioral responses to drugs of abuse (73). Likewise, similar results were demonstrated in self-administering rats receiving intra-NAc infusions of SAHA, where HDAC inhibition resulted in an upward shift in the dose response curve (FR schedule) and an increase in the break point under a progressive-ratio schedule. These data indicate that intra-NAc HDAC inhibition enhances motivation for self-administered cocaine (74).
Histone H3 phosphorylation and phosphoacetylation, both of which are considered activating marks of transcription, play key roles in the development of repeated drug-induced behaviors. Cocaine exposure rapidly induces H3 phosphorylation and phosphoacetylation globally (i.e. differentiation between striatal cell-types was not determined) in the striatum (65, 75). The H3 kinase mitogen- and stress-activated protein kinase (MSK1), which is a downstream member of the MAP kinase–signaling cascade, directly mediates cocaine-induced H3 phosphorylation; likewise, knockout of MSK1 results in reduced locomotor responses to the drug (75). Interestingly, loss of MSK1 prevents the acute induced expression of the immediate early gene (IEG) c-fos, the expression of which is induced in response to acute cocaine exposure and is decreased in response to chronic cocaine (an event that is potentiated by HDACis) (65, 75–77). Such decreased expression of c-fos transcription results, in part, from recruitment of a co-repressive complex containing ΔFosB and HDAC1 at the c-fos promoter (77). Because acute behavioral responses to cocaine require increased striatal activity, an electrophysiological response that is known to induce c-fos, it is quite intriguing that loss of MSK1, which reduces cocaine-induced H3 phosphorylation and locomotor sensitivity, results in the loss of cocaine-mediated c-fos transcriptional activity. Furthermore, MSK1-induced H3 phosphorylation can be enhanced via glutamate treatment, suggesting a possible role for such regulation in the development of synaptic plasticity (78).
Although strong evidence suggests a role for more transient histone modifications in the regulation of acute and chronic drug-induced behaviors, only recently has the more stable modification of histone methylation been proposed as an essential regulator of drug associated transcriptional, behavioral, and synaptic plasticity (7). Following a genome-wide promoter analysis of histone modifications after chronic cocaine exposure, not only were histone acetylation patterns altered by repeated cocaine administration, but a specific histone methylation mark, histone 3 lysine 9 di-methylation (H3K9me2), was shown to be strongly regulated at a number of gene promoters by cocaine (79). To further examine these findings, a transcriptional profiling approach was employed to identify whether specific histone methylating or demethylating enzymes may be altered in their levels of gene expression following repeated cocaine exposure. Only two enzymes, G9a [(also termed euchromatic histone-lysine N-methyltransferase (Ehmt2)] and G9a-like protein (GLP/Ehmt1) displayed persistently decreased expression, following both repeated EA and SA cocaine administration, suggesting a potential role for these enzymes in the development of cocaine-induced behaviors (7). Both G9a and GLP are ubiquitously expressed lysine methyltransferases (KMTs) that specifically catalyze mono- and di-methylation of H3K9 (H3K9me1 and H3K9me2) (80). This mark is highly, positively correlated with transcriptional repression and is found primarily throughout euchromatic regions of the genome. Consistent with decreased expression of G9a and GLP following chronic cocaine treatment, global levels of H3K9me2 was also shown to be reduced in the NAc (7). As mentioned earlier, many alterations in cocaine-induced gene expression appear to be rather transient following drug administration, yet numerous genes become more transcriptionally inducible (i.e., sensitized or primed for immediate transcription) following repeated drug exposure, a phenomenon largely dependent on G9a expression in the NAc. Furthermore, NAc-specific knockout of G9a, thus mimicking the effects of repeated cocaine exposure, led to an enhancement of drug-associated reward behavior, whereas overexpression of G9a blunted this response (7). In an attempt to identify the mechanisms through which G9a is repressed by chronic cocaine exposure, as well as to identify putative G9a target genes regulated by the drug, a negative feedback-loop was uncovered in which, under normal conditions, G9a acts to maintain homeostatic levels of gene expression in the NAc; with chronic treatment of the drug, however, such recruitment is diminished, either through repression of G9a or loss of G9a activity, leading to the enhanced inducibility of numerous synaptic plasticity-related transcripts. This process is ΔFosb-dependent, whereby following an acute dose of cocaine, G9a binding to the fosB promoter is dramatically increased, thus rapidly repressing cocaine-induced increases in ΔFosB expression; however, following repeated cocaine exposure, ΔFosB accumulation leads to G9a repression and reduced levels of H3K9me2 in the NAc, thus preventing G9a’s ability to maintain basal levels of gene expression in this brain region, ultimately enhancing behavioral responses to the drug (7). This inability to repress synaptic plasticity–related gene expression following chronic cocaine treatment results in enhanced dendritic spine plasticity in the NAc, a phenomenon positively correlated with increased behavioral sensitivity to the drug (6, 7). Indeed, NAc specific knockout of G9a, which dramatically increases animals’ CPP for cocaine, significantly enhances dendritic spine density on MSNs in the NAc, whereas viral overexpression of G9a, which reduces behavioral responses to cocaine, prevents the induction of cocaine-mediated increases in spine density in this brain region (7). Furthermore, ΔFosB overexpression in the NAc, in the absence of cocaine, is sufficient to reduce G9a recruitment to many of these plasticity-related gene promoters, and ΔFosB was further demonstrated to be both necessary and sufficient for cocaine-induced increases in MSN spine density in this brain region (7). These findings mark the first evidence of a role for chromatin regulation in the structural/morphological changes contributing to drug-induced behavioral sensitivity following repeated administration.
Transcriptional Mechanisms Underlying Drug-induced Structural Plasticity
Although it is clear that the G9a-ΔFosB negative feedback loop described above controls cocaine-induced increases in spine density, there is no evidence that G9a or ΔFosB can directly regulate the actin cytoskeleton, which has spurred a series of studies to uncover the downstream transcriptional targets of G9a-ΔFosB. Several recent studies have characterized candidate genes downstream of ΔFosB or G9a that are likely to be involved in this form of synaptic remodeling. Through the use of genome-wide analyses, ΔFosB overexpression was found to regulate several genes previously implicated in spinogenesis, including Cdk5 and MEF2: induction of Cdk5 leads to increased phosphorylation and inhibition of MEF2, which in turn increases dendritic spines on NAc MSNs (9). Subsequent evidence suggests that another transcription factor, NF-κB, is involved in positively regulating spinogenesis following chronic cocaine exposure. Both acute and chronic cocaine induce NF-κB activity in the NAc, resulting in the activation of NF-κB-dependent signaling cascades necessary for dendritic spine formation on MSNs (10). As with the Cdk5-MEF2 pathway, ΔFosB is required for cocaine-mediated transcription of NF-κB subunits, indicating that ΔFosB regulates an extensive program of aberrant gene expression resulting in increased spinogenesis on NAc MSNs. ΔFosB, dk, and the NF-κB subunits are transcriptionally regulated, in part, through a histone methylation–dependent mechanism following chronic cocaine administration, whereby repressive H3K9 methylation is reduced at these genes’ promoters, resulting in their enhanced transcriptional inducibility (7, 10). ΔFosB accumulation following chronic cocaine exposure decreases the expression and activity of G9a, and such reduced G9a activity leads to decreased recruitment of G9a to the cdk5, nfκb, and fosb promoters, resulting in a more permissive chromatin state (7). In this active state, numerous cofactors are recruited to these gene promoters—including histone acetyltransferase (HATs), histone kinases and attendant phosphatases, and histone KMTs—which promote enhanced gene expression of these structural plasticity–related genes (Figure 3). Although numerous groups have shown that these molecular mechanisms are necessary for drug-induced structural plasticity, none of these molecules can regulate actin polymerization directly, adding an additional layer of complexity to this pathway. It is conceivable that activation of these transcription factors increases gene expression of actin cytoskeleton-regulatory genes such as the neural Wiskott-Aldrich syndrome proteins (N-WASP) and WAVE proteins, both of which have been identified as having putative MEF2 and NF-κB binding sites in their proximal promoter regions (9). Additionally, WAVE1 regulates spine morphogenesis in a Cdk5-dependent manner (81, 82), and in this way, Cdk5 activation, induced by chronic cocaine via ΔFosB, could result in regulation of WAVE activity, whereas MEF2 or NF-κB may regulate the expression of WAVE proteins to further mediate long-term changes involved in the development of drug addiction. These and many other explanations are currently being tested.
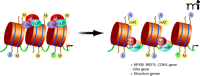
Cocaine induces chromatin modifications on gene promoters. As stated in Figure 2, methylation of histone 3 Lysine 9 (H3K9me2) causes chromatin condensation and gene repression. Under normal conditions, a complex of histone methyltransferases (HMTs) G9a, GLP, and histone deacetylases (HDACs) binds to histones and represses transcription. Following chronic cocaine administration, there is reduced binding of KMTs and HDACS, which, in combination with increased binding of histone acetyltransferases (HATs), ΔFosB, and RNA polymerase lI (POL II), results in a permissive state of chromatin and increased structural plasticity-related gene expression in NAc medium spiny neurons.
Functional Relevance of Drug-induced Structural Plasticity
Although we are starting to learn more about the transcriptional mechanisms driving structural plasticity, the functional relevance in promoting addictive behavior is far less clear. For example, inhibition of Cdk5 or activation of MEF2 (both of which would be expected to oppose cocaine’s effects) paradoxically enhance behavioral responses to cocaine (9, 63, 83). These unexpected findings suggest that gross changes in spine density may not lead to sensitized drug responses, per se, but may be a result of “homeostatic adaptations,” which compensate for additional changes caused by chronic cocaine exposure, such as a reduction in glutamatergic stimulation of MSNs via prefrontal cortical afferents (13, 84). On the other hand, direct inhibition of the ΔFosB and NF-κB pathways using viral gene transfer or conditional transgenic mice expressing dominant negative mutants of these proteins, decreases behavioral responses to cocaine, in line with the prevailing hypothesis that cocaine-induced increases in spine density mediate behavioral sensitization (7, 10). Furthermore, inhibition of G9a activity, which directly results in enhanced plasticity-related gene expression, amplified ΔFosB and NF-κB expression, and increased MSN dendritic spine density, thereby promoting increased sensitivity to the behavioral effects of chronic cocaine (7). The paradoxical differences between the behavioral effects of Cdk5-MEF2 as compared to those effects of NF-κB—despite the fact that induction of both pathways is mediated via G9a and ΔFosB, and that both pathways lead to increased dendritic spine density—highlight the complexity of these intracellular pathways and the importance of future research. Our hypothesis is that the net effect of cocaine is to induce, via G9a and ΔFosB, NAc spine density through multiple downstream targets (e.g., NF-κB, Cdk5-MEF2, many others) and that the net behavioral consequence of such signaling results in sensitized behavioral responses to cocaine (Figure 3). At the same time, however, an individual target pathway like Cdk5-MEF2 may, in isolation, elicit distinct behavioral effects via its own diverse downstream molecular consequences. Thus, it is crucial that future studies profile the downstream molecular pathways for numerous cocaine and ΔFosB targets to gain further insight into the specific contributions of each pathway to cocaine-induced spinogenesis and subsequent heightened behavioral responses to cocaine. We must also consider that by measuring the number of dendritic spines only, we are losing vital information about whether these new spines actually form functional synapses. Based on these studies, it appears that gross spine density changes do not drive or predict behavioral sensitization to cocaine, suggesting that a more detailed analysis of structural plasticity is needed (85).
Conclusions
The transcriptional mechanisms induced by chronic cocaine to regulate synaptic and structural plasticity are highly complex and require enzymatic reactions leading to permissive states of gene transcription. In this review, we have highlighted some recent advances in our understanding of these transcriptional mechanisms that require induction of the transcription factor ΔFosB. A novel mechanism of transcriptional regulation has been identified which utilizes the KMT G9a and drives a complex transcriptional profile, leading to alterations in synapse structure and addictive behavior. These changes may partly explain the very long-lasting behavioral plasticity that marks the chronic “addicted” state and points to the possibility of developing highly novel therapeutics to treat addiction by targeting enzymes that modify chromatin and thus transcriptional events responsible for relapse. Although more work is needed, we believe that the combined approaches discussed here to identify highly complex transcriptional profiles of the “addicted” state, along with a detailed analysis of synaptic plasticity, provides us with a unique perspective on the potent effects abused drugs have on cellular function.
- Copyright © 2010
References

Ian Maze, PhD, is a recent graduate from the the Mount Sinai School of Medicine where he worked in the laboratory of Eric J. Nestler. His research interests center on the molecular and biochemical underpinnings of activity-dependent regulation of chromatin dynamics in adult brain.

Scott Russo, PhD, is an Assistant Professor of Neuroscience at the Mount Sinai School of Medicine. His work focuses on the molecular mechanisms that control synaptic plasticity of reward circuitry in stress- and addictive-disorders. E-mail scott.russo{at}mssm.edu; fax 212-849-2611.

