The Biology of Caveolae: Lessons from Caveolin Knockout Mice and Implications for Human Disease
Abstract
Caveolae, plasma membrane invaginations that serve as membrane organizing centers, are found in most cell types, but are enriched in adipocytes, endothelial cells, and myocytes. Three members of the caveolin family (Cav-1, -2, and -3) are essential for the formation of caveolae. Specialized motifs in the caveolin proteins function to recruit lipids and proteins to caveolae for participation in intracellular trafficking of cellular components and operation in signal transduction. Mutations in the gene encoding CAV-1 are associated with the development and progression of breast cancers, whereas mutations in the CAV-3 gene result in Rippling Muscle Disease and a form of Limb-Girdle Muscular Dystrophy. The generation of caveolin-null mice has confirmed the essential role of these proteins in caveolae biogenesis and in the pathophysiology of diverse tissues. Caveolin-null mice provide new animal models for studying the pathogenesis of a number of human diseases, including cancer, diabetes, atherosclerosis, restrictive lung disease and pulmonary fibrosis, cardiomyopathy, muscular dystrophy, and bladder dysfunction.
Introduction
Morphologically distinct plasma membrane invaginations were first described in 1953 by George Palade from electron micrographs of endothelial cells (1). Yamada described similar structures in the gall bladder epithelium in 1955, and coined the term caveolae to reflect their appearance as “little caves” (2). Characterized as 50–100 nm flask-shaped invaginations at the plasma membrane, caveolae lack an electron dense coat (3). The elucidation of the structure of clathrin-coated vesicles in 1975 gave rise to the field of clathrin-mediated endocytosis (4), which served as a model for vesicular trafficking; however, the literature regarding caveolae remained limited by the lack of adequate biochemical markers (5, 6). In 1989, Glenney first identified caveolin as a 21–22-kDa tyrosine-phosphorylated substrate in chick fibroblasts transformed by the tyrosine kinase–encoding v-Src oncogene (7). Subsequent research identified caveolin as a protein component of caveolae (8) and as an integral membrane protein in trans-Golgi–derived vesicles (9).
The recent identification of a family of three related caveolin genes (CAV-1, CAV-2, and CAV-3) has enabled researchers to probe the biochemical properties and mechanisms of cellular trafficking of caveolae. To date, most of the research on caveolins has elucidated their structural role in caveolae biogenesis and their physical and functional interactions with proteins involved in cell signaling (10). The generation of genetically altered mice deficient in various caveolin proteins has provided an excellent opportunity to examine the functional consequences associated with the loss of these proteins and their impact on caveolae.
Caveolin Gene Family
Three members of the caveolin gene family have been identified (Figure 1⇓). Caveolin-1 (Cav-1) was identified as a tyrosine-phosphorylated protein in Rous sarcoma virus–transformed fibroblasts (7) and later was determined to be a structural component in caveolae (8). CAV-1 is composed of three exons that are highly conserved in sequence across species. Two isoforms have been identified, the predominant 178-residue Cav-1α, and the 147-residue Cav-1β derived from an internal translation initiation site (Met32) (11). Caveolin-2 (Cav-2) was identified in caveolae-enriched adipocyte membranes. CAV-2 is composed of three exons that encode a 162-residue protein whose sequence is highly similar to that of Cav-1. Cav-2α represents the full-length protein; two additional truncated isoforms (Cav-2β and Cav-2γ) have been identified, but remain uncharacterized (Figure 1B⇓) (12). CAV-3 was identified through database searches and analyses of cDNA libraries for genes homologous to CAV-1. It is composed of two exons that encode a single 151-residue mature protein (13). All three genes in mouse are located on chromosome 6, and the analogous boundaries between the last exons in CAV-1 and CAV-2 suggest they arose from gene duplication (14) ; however, Cav-3 and Cav-1 proteins share the greatest amino acid sequence identity. All three proteins contain a signature structural motif (FEDVIAEP) that is conserved across species (Figure 1B⇓) (10). The cell-specific expression patterns of Cav-1 and Cav-2, which normally colocalize, are distinct from that of Cav-3, which is muscle-specific (13, 15).
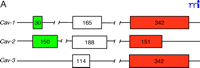
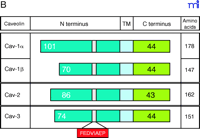
The caveolin gene family. A. The exon arrangement in Cav-1, Cav-2, and Cav-3 is indicated by boxes; the number of nucleotides in each exon is shown. B. The three caveolin proteins (Cav-1, Cav-2, and Cav-3), including the two isoforms of Cav-1, can be depicted in terms of N-terminal, transmembrane (TM), and C-terminal domains. The position of the conserved FEDVIAEP signature motif is illustrated.
The human CAV-1 and CAV-2 genes map to chromosome locus 7q31.1 near the D7S522 genetic marker. D7S522 encompasses a known fragile site (FRA7G) within 7q31.1 (16). A large number of epithelial cancers (e.g., breast, prostate, ovarian, and renal) have deletions distributed around the D7S522 marker (17 –19), suggesting that a tumor suppressor gene resides within this region. Although no genes have been directly mapped to this locus, the closest known gene to this region is CAV-2, located just 67 kb downstream; CAV-1 resides an additional 19 kb further downstream (20). Accumulating evidence implicates CAV-1 as the tumor suppressor gene disrupted at D7S522 (21). Indeed, sixteen percent of human breast cancer samples examined (i.e., invasive carcinomas) have CAV-1 mutations (22). A specific Cav-1 point mutation produces a protein (i.e., P132L) that acts in a dominant-negative manner and is sufficient to transform mouse fibroblasts (NIH 3T3) in vitro (17, 22). Furthermore, the GC-rich region of the CAV-1 promoter is hypermethylated in breast cancer cell lines and prostate tumor samples, suggestive of transcriptional silencing (20, 23) ; however, another report did not detect a change in the methylation status of CAV-1 in human tumor cell lines (24). These disparate conclusions may reflect the different biochemical methods used to detect the status of cytosine methylation. Additional work will be needed to clarify the role of methylation in the regulation of CAV-1 gene expression.
Interestingly, a recent report suggesting the Suppression of Tumorigenicity-7 (ST7) gene as the putative tumor suppressor gene located within the 7q31 region has been refuted by subsequent research. Mutations in the ST7 gene were initially reported in human breast and colon carcinomas, and the expression of the ST7 cDNA in human PC3 prostate tumor cells inhibits their tumorigenicity in nude mice (25). However, several reports have since repudiated these data (26–28).
Human CAV-3 maps to 3p25 (29, 30), approximately 7–10 kb from the oxytocin receptor gene near D3S18, the genetic marker of two known diseases, von Hippel-Lindau disease and 3p-syndrome (31). CAV-3 disruption may be involved in 3p-syndrome, which results from a hemizygous deletion of 3pter–p25 and is characterized by growth retardation, specific craniofacial features (i.e., microcephaly, ptosis, and micrognathia), mental retardation, and cardiac septal defects (32). Several specific mutations have been identified in CAV-3 that lead to a type of limb-girdle muscular dystrophy (discussed below).
Structural Domains of the Caveolin Proteins
Cav-1 lacks an N-terminal signal sequence but contains a single hydrophobic domain (residues 102–134) that is required for its cotranslational insertion within the endoplasmic reticulum (ER) (33), whereby both the N and C termini of Cav-1 remain cytoplasmic (Figure 2⇓). Both N and C termini possess membrane attachment domains (N-MAD, residues 82–101; and C-MAD, residues 135–150). The N-MAD domain directs Cav-1 to caveolae membranes, whereas C-MAD directs localization to the trans-Golgi network (34). Three residues (i.e., Cys133, Cys143, and Cys156) in the C-terminal domain are palmitoylated and stabilize Cav-1 structure at the membrane, but are not essential for membrane binding or caveolar localization (35, 36). Cav-1 forms homo-oligomers through its oligomerization domain (i.e., residues 61–101) (37, 38), but oligomerization between Cav-1 and Cav-2 might be mediated by their membrane spanning domains (15). Given the robust colocalization of Cav-1 and Cav-2 proteins, hetero-oligomers of 350-kDa (composed of approximately fifteen monomers) are favored in situ (39). These large structures form rapidly after synthesis of Cav-1 in the ER and before transport to the Golgi apparatus (33). Cav-2 is incapable of forming large homo-oligomers, but exists as monomers or dimers that are retained in the Golgi apparatus in the absence of Cav-1 (40, 41). Hence, Cav-2 is dependent on Cav-1 for caveolar localization, consistent with their extensive coregulation and common tissue-specific expression (42). Cav-3 forms homo-oligomers in situ and does not bind Cav-2 (13).
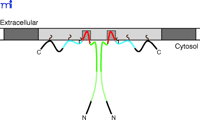
Topology of caveolin at the inner cell membrane. A dimer of Cav-1 is represented; interprotomeric association is maintained through the so-called oligomerization domain (dark and light green). Association of Cav-1 with the cell membrane is maintained by N-terminal and C-terminal membrane attachment domains (dark green and blue, respectively), the transmembrane domain (red), and three palmitoyl groups (brown). The plasma membrane is indicated in dark gray; cholesterol- and sphingomyelin-enriched membrane is indicated in light gray.
Caveolae Organelles
The plasma membrane exists as a lipid bilayer composed of both liquid-disordered and liquid-ordered domains. In the liquid-disordered state, the phospholipids are capable of rapid lateral diffusion. However, liquid-ordered domains confer a more rigid bilayer assembly, with confined movement of the phospholipids that results from the coalescence of cholesterol, glycosphingolipids and sphingomyelin into “lipid rafts” (43). These domains are resistant to detergent solubilization and have increased buoyancy compared to other membrane phospholipids, a characteristic that has allowed their purification by sucrose gradient ultracentrifugation (44). These biochemical properties are also shared by caveolae, but caveolins are localized selectively to caveolae. Cholesterol is essential for caveolae formation, and Cav-1 is a cholesterol-binding protein (8, 45, 46). Cav-1 transcription is activated by cholesterol but repressed by oxysterols (47, 48). Indeed, there is a cellular threshold of cholesterol needed for caveolar biogenesis, a requirement that also exists for the formation of clathrin-coated and synaptic vesicles (49 –51). Cholesterol-depleting agents result in the loss of morphologically identifiable caveolae, so that Cav-1 is associated with a flattened membrane before being lost from the membrane surface (8).
Adipocytes appear to have the highest concentration of Cav-1 and Cav-2 and the largest number of caveolae (52). In murine fibroblastic 3T3-L1 cells, differentiation into adipocytes increases the number of caveolae approximately ninefold, and the expression of Cav-1 and Cav-2 rises by about twentyfold (53) ; Cav-1 is a major fatty acid–binding protein in adipocytes (54). These data suggest that Cav-1 and caveolae act in concert with other proteins as portals for the uptake and transport of fatty acids into lipid droplets (55, 56). Although lipid droplets are most prevalent in adipocytes, they also exist, to varying extents, in most cells (57). These droplets consist of a core of neutral lipids (e.g., triglycerides and cholesterol esters) encased in a phospholipid monolayer derived from the ER. After their transport through the ER, Cav-1 and Cav-2 can also be transported to lipid droplets (55, 58). Surprisingly, the specific overexpression of Cav-2β results in the constitutive localization of Cav-2β to the surface of lipid droplets (59), suggesting a role for this Cav-2 isoform in lipogenesis.
Heterologous overexpression of Cav-1 in cells lacking endogenous Cav-1 results in the production of caveolae, whereas targeted downregulation of Cav-1 in cells results in the loss of caveolae (60, 61–63). Hence, Cav-1 is essential for the process of caveolar biogenesis (Box 1). Based on these data, a model for caveolae biogenesis may be imagined in which the strong binding of Cav-1 to cholesterol and sphingolipids in lipid rafts, along with large multimeric Cav-1 complexes and the cytoskeleton, produces the structural forces necessary for the characteristic bending of the plasma membrane (10). Indeed, a C-terminal sequence of Cav-1 is able to interact with full-length Cav-1 and, consequently, form higher-order oligomeric complexes by adjoining respective C-terminal domains (37, 64). Cav-2 expression alone is insufficient to induce caveolae, and in the absence of Cav-1, the Cav-2 protein is restricted to the Golgi apparatus (42).
Caveolae Facts: Biogenesis and Morphology
Cholesterol is required for caveolar biogenesis, as its depletion by filipin and methyl-β -cyclodextrin ablates caveolae.
Cav-1 is a cholesterol-binding protein, interacting with plasma membrane cholesterol and thereby driving caveolar formation.
Downregulation of Cav-1 by antisense reduces the number of caveolae at the plasma membrane.
Overexpression of Cav-1 in cells lacking endogenous expression induces caveolae biogenesis.
Cav-1-null and Cav-3-null mouse tissues and cultured cells lack caveolae as normally seen in wild-type animals and cells.
Caveolae are aligned along actin filaments, which copurify with caveolar membranes. Both myosin subfragment I and the actin cross-linking protein filamin have been identified as ligands for Cav-1 (44, 65). Filamin interacts with the N-terminal domain of Cav-1 and is colocalized to patches of Cav-1 at the plasma membrane. The Cav-1β isoform, which lacks 31 N-terminal amino acids compared to Cav-1α, has a higher affinity for filamin (66). Additional proteins, such as inositol-1,4,5-trisphosphate receptor-like proteins and calcium channels, localize to caveolae and may facilitate anchorage to actin filaments (76). Although its significance is unclear, the binding of Cav-1 to the cell cytoskeleton may reflect both an organizational framework for caveolae distribution and serve as a necessary anchorage point for caveolae formation at the plasma membrane. Microtubules and their associated motor proteins may be involved in the intracellular movement of Cav-1–bearing vesicles (68).
Endocytosis
The process of endocytosis involves the engulfment of extracellular substances at the plasma membrane and their internalization by free membrane vesicles that bud from the surface and are selectively targeted to intracellular membrane compartments. This process involves multiple mechanisms and a variety of vesicular trafficking pathways. Clathrin-coated pits represent the prototypical cell system for the endocytotic process (69). Caveolae are thought to participate in a parallel endocytotic pathway to that mediated by clathrin-coated vesicles. Indeed, the structure of caveolae implies a dynamic process of budding from the membrane surface; nevertheless, they exhibit a uniform morphology across the plasma membrane, with no distinct intermediary stages associated with vesicle budding, such as structures often seen with clathrin-coated pits (70).
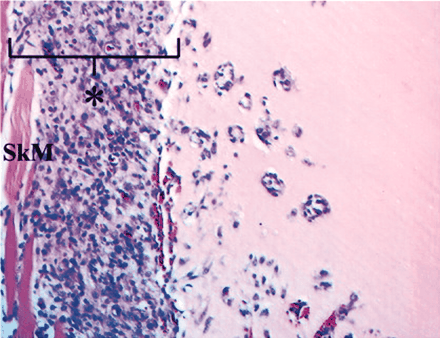
It is unclear where internalized caveolar vesicles are intracellularly routed. There may be several strategies that include transit to the trans-Golgi, ER, endosomes, lysosomes, and larger caveosomes (i.e., distinct intracellular Cav-1–positive membrane compartments that could be derived from budded caveolae or directly targeted from the ER/Golgi; see Figure 3⇓) (68, 71). Thus, Cav-1–containing vesicles appear to selectively traffic cargo proteins, which suggests that different Cav-1–bearing vesicles contain unique complements of proteins that trigger caveolae endocytosis and directional transport to specific intracellular compartments (72). There appear to be at least three major Cav-1–containing compartments: 1) plasma membrane caveolae immobilized by cortical actin; 2) Cav-1–positive vesicles, termed “cavicles,” that move along microtubules; and 3) large pericentrosomal caveosomes (68, 73, 74). These Cav-1–containing structures may be further subdivided into unique cell-specific compartments. For example, endothelial cells contain large Cav-1–positive vesiculo-vacuolar organelles (VVOs) that appear as clusters of caveolae-like vesicles that originate at the membrane surface and penetrate into the cytosol (Figure 3⇓) (75). In addition, endothelial cells in skeletal muscle possess open caveolae, whereas lung alveoli contain closed caveolae that possess a non-membranous diaphragm that caps the caveolar opening (Figure 3⇓) (76). A protein component, termed plasmalemma vesicle protein-1 (PV-1), was recently cloned and identified as a structural element of the caveolar diaphragm (77). Both the caveolar diaphragm and the PV-1 protein are localized to some transendothelial channels and fenestrae (78). This diaphragm has also been observed in VVOs, and the PV-1 protein is localized to a diverse array of cell types that are of endothelial and endocrine origin (79). The PV-1 protein represents an interesting biochemical link between many of these endothelial membrane structures and caveolae, but its functional significance remains unknown. The importance of these various membrane compartments and there dependency on Cav-1 is only now being elucidated.
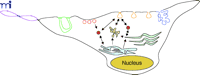
Intracellular trafficking of caveolae-related compartments. The following subcellular membrane compartments are shown: fenestrae (violet), channels (green), plasmalemma vesicles (red), caveolae (orange), vesiculo-vacuolar organelles (blue), caveosomes (yellow), and cavicles (solid rust circles). Arrows depict the movement of caveolae-derived vesicles (orange) from the cell surface to caveosomes (yellow), the Golgi apparatus (light green), and the ER (light blue).
The diverse array of Cav-1–containing compartments may reflect the multiplicity of cell functions mediated by these membrane-organizing domains. Although several mechanisms allow ligand-triggered internalization in Cav-1–associated vesicles, caveolae themselves may function more in membrane topology, serving to organize the plasma membrane into different functional units. The selective localization of many signaling molecules to caveolae illustrates the importance of this microdomain for cell responses to exogenous stimuli and highlights the ability of caveolae to mediate diverse events through their ability to recruit cell-specific proteins
.
Cell Signaling
The functional role of caveolae in cell signaling has been covered extensively in several recent reviews (10, 80). The “caveolae signaling hypothesis” is a general model to explain the behavior of caveolae-mediated signaling events. It states that caveolae function to compartmentalize various signaling molecules and, consequently, serve as organizing centers, selectively recruiting or sequestering molecules (81, 82). Furthermore, caveolae provide a structural platform by which regulated changes in the activation state of a given sequestered molecule can occur rapidly in response to extracellular stimuli.
A plethora of signaling molecules copurify with Cav-1 in isolated caveolae-derived membrane fractions (10), and data suggest that there is a direct physical interaction between many of these signaling molecules and Cav-1 (83, 84). A 20-residue hydrophobic scaffolding domain (Cav-1 residues 82–101) interacts with a variety of signaling proteins [e.g., H-Ras, c-Src, insulin receptor, endothelial nitric oxide synthetase (eNOS), Gα subtypes] to form catalytically inactive complexes (85, 86). Interaction of the Cav-1 scaffolding domain (CSD) with caveolin-binding domains (CBD) in other proteins can result in the inhibition of normal downstream signaling mediated by the sequestered protein. One of the best studied examples is eNOS, which is tonically inactivated by its interactions with Cav-1 (87). Following an appropriate stimulus, Cav-1–mediated repression is released (disinhibition) and the propagation of downstream signaling occurs (43, 88).
Cav-1 in Cell Transformation
The action of Cav-1 as a tumor suppressor is based on its capacity to inhibit the signaling activity of several protooncogene products that impart growth and survival advantages to the cell (89). The first implication of Cav-1 in cancer was the observation that Cav-1 is tyrosine-phosphorylated in v-Src–transformed fibroblasts (7), followed by the observation that oncogene-mediated transformation of murine NIH 3T3 fibroblasts results in the transcriptional downregulation of Cav-1 and a loss of identifiable caveolae organelles (90). Additionally, decreasing the intracellular concentration of Cav-1 through the use of an antisense cDNA approach is sufficient to cause the cellular transformation of NIH 3T3 cells (91). The reduction in Cav-1 and caveolae appears to be a common event in transformed cell lines, suggesting that Cav-1 might be “inactivated” during tumorigenesis (92, 93). Indeed, numerous oncogenes abrogate the transcription of Cav-1 (62, 90, 93), but the exact mechanism of this transcriptional repression remains to be determined. Hypermethylation may provide a common mechanism, but both c-Myc and p53 control Cav-1 transcription through distinct promoter elements (92, 94).
Interestingly, cell transformation mediated by an activated H-Ras mutant (i.e., G12V) decreases Cav-1 protein levels and promotes anchorage-independent cell growth. Conversely, the overexpression of Cav-1 in H-Ras G12V–transformed cells inhibits anchorage-independent growth, reverting the transformed cell phenotype (62). Loss of cell adhesion is a hallmark of cell transformation, as normal cells usually undergo apoptosis in response to loss of cell attachment (95). Cav-1 expression correlates with a decrease in cell motility, and phosphorylation of Cav-1 on Tyr14 results in its localization to focal adhesions (96, 97). Although the role of Cav-1 at focal adhesions remains enigmatic, it likely serves to recruit and organize proteins and lipids necessary for substrate attachment. Cav-1 localized at focal adhesions is thought to bind c-Fyn and a subset of β 1 integrins that signal through Shc, which is required for fibronectin adhesion leading to activation of the Ras–p42/44 MAP kinase cascade (98, 99).
These data suggest that the loss of Cav-1 disrupts cellular adhesion and consequently can either induce apoptosis when proper cell cycle checkpoints are in place or promote metastasis when synergistically combined with uncontrolled cell growth. The role of Cav-2 and Cav-3 in cell transformation is unknown, but cholesterol depletion or expression of a dominant-negative Cav-3 protein is known to inhibit Ras-mediated signaling (100).
Multidrug resistance (MDR) is defined as cellular resistance to a divergent group of structurally and functionally distinct drugs and thus limits the efficacy of chemotherapeutic strategies used to treat cancers (101). Many cellular changes accompany the MDR phenotype, including changes in membrane lipid composition and caveolae (102). Often, MDR cells express high levels of both Cav-1 and Cav-2, with a high caveolae surface density (103, 104). P-glycoprotein and related ATP-dependent drug efflux pumps mediate cellular drug resistance by actively extruding drugs from cells (105). Additionally, alterations in the pharmacokinetics of drug uptake, metabolic detoxification, and cellular repair mechanisms can support MDR (101). The P-glycoprotein associates with caveolae, and in MDR cells, cholesterol, Cav-1, and raft-associated lipid levels are all increased (106). Indeed, overexpression of the P-glycoprotein in human colon and breast cancer cells increases Cav-1 levels, with a concomitant increase in membrane caveolae (104). Thus, in MDR, the upregulation of Cav-1 may accelerate the intracellular cholesterol efflux pathway thereby facilitating cytotoxic drug transport to membrane domains capable of clearance or neutralization (107).
Although Cav-1 may support MDR, increased Cav-1 levels can mitigate the transformed phenotype by promoting cell adhesion and senescence. Indeed, Cav-1 expression is inversely correlated with that of cyclin D1, a positive regulator of cell growth, in transformed cells (108). Furthermore, several MDR cell lines exhibit reduced tumorigenicity in athymic mice and reduced anchorage-independent cell growth.
Cav-1–/– mice do not form spontaneous tumors, but they exhibit an increase in tumorigenicity following exposure to carcinogens, as compared to their wild-type counterparts (109, 110). Repeated dermal applications of the carcinogen 7,12-dimethylbenzanthracene (DMBA) increases the incidence, number, and size of skin tumors in Cav-1–/– mice (Figure 4⇓) (110). Tumor formation is preceded by epidermal hyperplasia of the basal and suprabasal cell layers, with commensurate increases in both cyclin D1 and phospho-ERK1/2 levels.
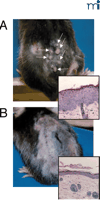
Tumorigenesis in Cav-1 −/− mice. Repeated dermal exposure to DMBA results in significantly higher rates of tumor formation (arrows) in Cav-1−/− mice (A), relative to wild-type mice (B). Insets demonstrate the increased epidermal cell hyperplasia of both the basal and suprabasal cell layers prior to tumor formation in animals that lack Cav-1.
A dominant-negative form of human CAV-1 (i.e., P132L) has been observed in many human breast cancers (91). Interestingly, the expression of the Cav-1 P132L mutant in vitro results in its retention in the Golgi apparatus. Coexpression of the P132L mutant along with wild-type Cav-1 results in the mistargeting of wild-type Cav-1 to the Golgi apparatus. Therefore, the P132L mutant Cav-1 serves as a dominant-negative modulator of wild-type Cav-1 by promoting its intracellular accumulation (17). Although Cav-1–/– mice do not show any increased incidence of spontaneous mammary tumor formation, they show significant mammary epithelial cell hyperplasia as early as six weeks of age, which is also accompanied by increased lobular development, with acini formation and fibrosis (17).
Tumor-prone transgenic mice harboring the gene that encodes the polyoma middle T antigen under the control of the mouse mammary tumor virus promoter (i.e., MMTV-PyMT) develop spontaneous multifocal mammary tumors at about eight weeks of age, with significant pulmonary metastases (111). When Cav -1–/– mice are crossed with the MMTV-PyMT line, the offspring show, at three weeks of age, a dramatic increase in the number, size, and grade of dysplastic mammary lesions (Figure 5⇓). Interestingly, this phenotype requires the loss of both Cav-1 alleles, as Cav-1 hemizygotes harboring the MMTV-PyMT construct do not show any increase in the formation of mammary dysplastic foci. Mechanistically, the increase in dysplastic mammary lesions associated with Cav-1–/– mice is concomitant with the upregulation of cyclin D1 (112). The phenotype of the Cav-1–/– mutation—that is, the increased sensitivity to carcinogens without an apparent tendency towards spontaneous tumor development—is consistent with our understanding of tumorigenesis as a multistep process that can involve the selective and progressive accumulation of oncogenic mutations. Indeed, the etiology of most cancers does reflect a series of cooperative insults to cell-cycle control (113). Because of its contribution to multiple signal transduction pathways, the obliteration of Cav-1 may be sufficient to potentiate transformation by disrupting downstream signaling events involved in cell-cycle regulation.
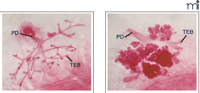
The influence of Cav-1 on mammary tumor foci in nulliparous transgenic mice. Three-week-old mice carrying the PyMT oncogene under the control of the MMTV promoter in a wild-type background (left) show significantly fewer foci relative to mice that lack Cav-1 (right). (PD, primary duct; TEB, terminal end buds.)
Physiological Roles of the Caveolins: Knockout Mice
Knockout mice that lack Cav-1, Cav-2, and/or Cav-3 are viable and fertile, which is remarkable, given the attributes of the caveolin proteins and their widespread tissue distribution. Perhaps the most striking observation is that mice deficient in both Cav-1 and Cav-3 have a complete loss of morphologically identifiable caveolae in both non-muscle and striated muscle tissues that normally express those proteins (109, 114). These animal models serve as definitive proof that both Cav-1 (non-muscle) and Cav-3 (muscle-specific) play an essential role in caveolar biogenesis. In contrast, Cav-2–deficient mice retain morphologically distinct caveolae. Hence, Cav-1 –/– and Cav-3 –/– mice provide unique models to study the tissue-specific loss of non-muscle and muscle caveolae, respectively. Although considerable progress has been made in delineating the phenotypic consequences of caveolin deficiencies in mice, the prospect of relating these observiations to human disease continues. Our current understanding regarding the phenotypes of caveolin-null mice are detailed below.
Cav-1 Knockout Mice
Prior to the generation of Cav-1 –/– mice, overexpression studies had implicated Cav-1 as a mediator of caveolae formation (41, 63). The generation of viable and fertile Cav-1–deficient mice was simultaneously reported by two independent groups (109, 114), both of whom described a complete absence of morphologically identifiable caveolae in all tissues that normally express the Cav-1 protein (i.e., endothelia and adipose) (109, 114–116), as well as in primary cell cultures established from the Cav-1 knockout mice (109). Cav-1 was thus unequivocally defined as essential for caveolae biogenesis in non-muscle cells (109, 114). Interestingly, striated muscle cells (i.e., cardiac and skeletal) that lack Cav-1 but express Cav-3 retain their caveolae.
Because the physicochemical process behind membrane invagination and the formation of caveolae remains unresolved, the consequence of Cav-1 loss may reflect either a primary functional loss or a secondary effect on membrane composition. As Cav-1 serves as a binding partner for lipids and proteins, its loss may prevent the organization of essential membrane components for caveolar biogenesis. Indeed, Cav-1–deficient cells have deficits in the sorting of both glycosylphosphatidylinositol-(GPI)-anchored and lipid-modified proteins such as the c-Src tyrosine kinase (117). Cav-1 also binds and transports cholesterol, so that its loss may alter cholesterol composition and organization within lipid rafts at the plasma membrane.
In Cav-1 –/– mice, Cav-2 protein levels are reduced to less than 10% of wild-type levels, but Cav-2 is not transcriptionally downregulated. Instead, it appears that the Cav-2 protein is dependent on Cav-1 for heteromultimeric association and trafficking to the plasma membrane (42). Cav-2 is unstable in the absence of Cav-1 and undergoes proteolytic degradation, so that only residual amounts remain trapped in the Golgi apparatus. Proteasomal inhibitors restore Cav-2 protein levels to normal and result in proper targeting to the plasma membrane, similar to the result seen when Cav-1 is reintroduced into Cav-1 –/– cells. Therefore, Cav-1 –/– mice are severely deficient in Cav-2, and the dramatic lung phenotype that they manifest appears to be the result of a Cav-2 deficiency (see below).
Metabolic Derangements
Neither the lipid composition of isolated lipid rafts nor the pattern of GPI-anchored proteins differs between wild-type and Cav-1 –/– mice (114). Cav-1 is a mediator of cholesterol transport (118, 119), but blood lipoprotein composition and cholesterol content of high-density lipoprotein (HDL) are apparently normal in Cav-1 –/– mice (114).
Adipose Abnormalities and Insulin Resistance
Intraperitoneal fat pads appear histologically normal in the Cav-1 −/− mice; however, young Cav-1 −/− mice totally lack the hypodermal fat layer, and female mammary adipocytes appear atrophic. Tissue-specific changes in adipose depots become more pervasive as these mice age. Cav-1 −/− mice are slightly hyperphagic but show a striking resistance to diet-induced obesity. Older Cav-1 −/− mice are lean, with clearly smaller fat pads than corresponding wild-type controls. In these older Cav-1 −/− mice, the adipocytes within the fat pads are poorly differentiated, with a decreased cell diameter (reflecting smaller lipid droplet size) and a hypercellular parenchyma (120).
Young Cav-1 −/− mice on a normal diet show insulin-resistance, whereas a high-fat diet results in postprandial hyperinsulinemia within nine months. The Cav-1 protein binds to the insulin receptor (IR-β) and functions as a positive regulator of receptor activation (121). Indeed, two mutations of the human insulin receptor (i.e., W1193L and W1200S, both within the established Cav-1–binding motif) result in severe insulin resistance (122, 123). Both mutations result in receptor destabilization, loss of autophosphorylation, and rapid degradation (124).
The adipose IR-β protein levels (but not corresponding mRNA levels) of Cav-1 −/− mice are reduced to less than 10% of wild-type values, so that IR-β destabilization and proteolytic degradation are likely consequences of Cav-1 deficiency. Indeed, the ectopic expression of caveolin-1 in Cav-1 −/− mouse embryonic fibroblasts (MEFs) completely restores IR-β expression to wild-type levels. Furthermore, a Cav-1 mutant protein that lacks the scaffolding domain (i.e., residues 61–100) is unable to rescue IR-β levels in these MEFs (125). Young Cav-1 −/− mice have elevated levels of triglycerides and free fatty acids, a condition exacerbated in the postprandial state. Analysis of lipid profiles shows a selective buildup of chylomicrons, likely the result of ineffective clearance of postprandial lipids by adipocytes (125). Hence, the lean phenotype of Cav-1 −/− mice may be attributed, at least in part, to a defect in insulin-mediated lipogenesis.
Premature Lactation
During lactation, Cav-1 expression in the mammary gland is downregulated, but its expression returns rapidly after weaning. That Cav-1 functions as a negative regulator of prolactin (PRL) signaling was first suggested by the Cav-1–mediated repression of PRL-induced β -casein transcription in cultured mammary epithelial cells (126). PRL binds to its receptor and activates the Jak-2/STAT5a pathway (127, 128). The scaffolding domain of Cav-1 includes a sequence very similar to the pseudosubstrate sequence of the suppressor of cytokine signaling (SOCS), a sequence crucial to the ability of SOCS to inhibit Janus kinases (Jaks); Cav-1 thus inhibits prolactin-induced STAT5a phosphorylation through a direct physical association with Jak-2 (129). Accordingly, Cav-1 −/− mice display accelerated development of the mammary lobuloalveolar system during pregnancy, leading to premature lactation. The accelerated lactation phenotype of Cav-1 −/− mice does not involve alteration of serum PRL levels, but rather is the result of hyperphosphorylation of STAT5a (i.e., the substrate of Jak-2 phosphorylation) (129).
Urogenital System Abnormalities
Urine calcium concentrations are significantly higher in Cav-1 −/− mice. At five months of age, approximately 67% of male Cav-1 −/− mice show urinary calcium stone formation; wild-type male mice develop urinary calculi at a rate of about 19% (130). The disruption of calcium absorption is likely due to the altered localization of the plasma membrane Ca-ATPase in distal convoluted tubules, which is normally localized to caveolae organelles.
Additionally, significant urinary bladder dysfunction and urogenital organ changes are observed in male Cav-1 −/− mice at twelve months of age (131), including a thickening of the urinary bladder smooth muscle layer with an associated increase in the ratio of urinary bladder-to-body weight. Cav-1 −/− mice show an increase in basal, threshold, and spontaneous urinary bladder pressures. In contrast, a reduction in the contractile response to both carbachol and KCl in is observed urinary bladder strips. These smooth muscle alterations are accompanied by significant fluid accumulation in the prostate and seminal vesicles, and intracellular vacuolization in the kidneys (131). The fibromuscular stroma of the prostate, which consists mainly of smooth muscle cells, is also hypercellular and thickened. In this regard, aging male Cav-1 −/− mice may provide a new animal model to study lower urinary tract dysfunction (LUTD), a common human disease, particularly for aging male patients.
Vascular System Dysfunction
Cardiac Hypertrophy
In the heart, Cav-1 is expressed in endothelial cells and fibroblasts, but not in myocytes; nevertheless, the loss of Cav-1 results in abnormal cardiac function. Quantitative imaging reveals an enlarged right ventricular cavity and a thickened left ventricular wall, with associated decreases in systolic function and myocyte hypertrophy and fibrosis of the perivascular spaces. In addition, Cav-1 −/− mice show hyperactivation of the p42/44 MAPK cascade in both cardiac tissue and isolated fibroblasts, along with increased eNOS activity (132). Interestingly, vascular smooth muscle cells (VSMCs) express all three caveolin proteins, and there is a surprising absence of caveolae in Cav-1 −/− mice, despite normal Cav-3 expression (114, 131)
Increased Production of Nitric Oxide
In the absence of Cav-1, eNOS activity does not respond to negative regulatory signals and consequently NO levels remain constitutively elevated. Isolated aortic rings from Cav-1 −/− mice do not maintain a constant contractile tone, oscillating at a frequency of once per second. The partially contracted quality of blood vessels, an important determinant of blood pressure that is coupled to changes in spontaneous transient outward currents (STOCs) and Ca2+ sparks (local increases in sub-sarcolemmal Ca2+), has been linked to caveolae. The frequency of STOCs in the VSMCs of Cav-1 −/− mice is significantly reduced at physiological membrane potential, consistent with elevated eNOS actvities (114). In addition, the vasodilatory response of aortic rings isolated from Cav-1 −/− mice is completely restored by the addition of the eNOS inhibitor, L-NAME (109).
Defects in Angiogenesis
Implants of Matrigel (a complex mixture of extracellular matrix components), administered to wild-type mice with the angiogenic agent FGF-2, are readily infiltrated by blood vessels; Matrigel plugs implanted into Cav-1 −/− mice show a significant reduction in blood vessel infiltration and vessel density (133). Similarly, subcutaneous injection of the melanoma cell line B16-F10 into Cav-1 −/− mice results in fewer and smaller primary tumors than result from wild-type animals; tumors that develop in the Cav-1 −/− animals, moreover, manifest decreased blood vessel density along with incompletely formed capillaries that lack caveolae. These data indicate that endothelial cells from Cav-1 −/− mice have a disrupted response to angiogenic growth factors (133).
Microvascular Hyperpermeability
Caveolae can mediate the endocytosis of bovine serum albumin (BSA) and facilitate its transcytosis across the endothelium (134, 135). Indeed, fluorescently labeled BSA is not taken up by cultured MEFs derived from Cav-1 −/− mice (109). Consistent with these observations, gold-conjugated BSA injected into the lung vasculature of Cav-1 −/− mice remains predominantly in the lumen, indicating the dysregulation of BSA transcytosis (Figure 7⇓) (136). Additionally, BSA uptake by aortic ring segments from Cav-1 −/− mice is severely compromised (136), although the in vivo clearance of iodinated BSA from the blood of Cav-1 −/− mice following vascular injection is faster than that of wild-type animals (137). These disparate results can be reconciled by the observation of morphological abnormalities in Cav-1 −/− endothelial cell tight junctions and defects in endothelial cell attachment to the basement membrane (137). Cav-1 −/− microvascular hyperpermeability can be normalized by treatment with the NO-synthase inhibitor L-NAME (137). Hence, caveolae-mediated transcytosis of BSA is lost in Cav-1 −/− mice, but there is increased microvascular hyperpermeability that can facilitate the paracellular movement of macromolecules, including BSA. This hyperpermeability may also explain why the concentration of BSA in the cerebral spinal fluid of Cav-1 −/− mice, a process thought to be mediated by caveolae, does not differ from that of wild-type mice (114). These data indicate that Cav-1, via its role in caveolae formation, is a positive regulator of transcytosis but conversely functions as a tonic inhibitor of eNOS that negatively regulates the paracellular movement of macromolecules.
Cav-1 in Atherosclerosis
Although the total plasma cholesterol level in Cav-1 −/− mice is normal, they exhibit basal hypertriglyceridemia (i.e., elevated VLDL/chylomicrons) that is not attributable to dysfunctional lipoprotein lipase activity (120). To investigate the contribution of Cav-1 to the development of atherosclerosis, Cav-1 −/− mice were interbred with atherosclerosis-prone knockout mice that lack apolipoprotein E (138), generating Apoe /Cav-1 double-knockout (dKO) mice. The loss of Cav-1 in the Apoe −/− genetic background results in significant elevations in plasma cholesterol and triglycerides, consistent with a lipoprotein profile that would promote the development of atherosclerotic lesions. Despite this “proatherogenic” lipoprotein profile, the loss of Cav-1 decreases atherosclerotic lesions by about 70% in the aortas of Apoe −/− mice. The dKO animals show a concomitant downregulation of the class B scavenger receptor CD36 and the adhesion molecule VCAM-1 (139). These phenotypes may reflect alterations in endothelial adhesion molecules that regulate monocyte recruitment and subsequent transmigration into the subendothelial space or, more likely, the decreased uptake/transcytosis of select lipoproteins by endothelia or macrophages.
The Effect of Cav-1 on Longevity
Cav-1 −/− mice manifest a significantly increased mortality rate, which peaks between twenty-seven and sixty-five weeks of age; hemizygous mice, retaining a single copy of the Cav-1 gene, are no more morbid than the wild type. The decreased lifespan of Cav-1 −/− mice appears to be secondary to progressive pulmonary fibrosis, hypertension, and cardiac hypertrophy that are apparent at twelve months of age in the form of thickened left and right ventricles, myocyte hypertrophy, and a significant decrease in left ventricular systolic function (140).
Cav-2 Knockout Mice
The generation of Cav-2 knockout mice has been accomplished by ablation of the gene’s first two exons, which results in the complete loss of all three isoforms of Cav-2 (115). Interestingly, Cav-2 −/− mice appear normal in terms of the expression and localization of Cav-1, the presence of caveolae, vascular tone, body weight, and lipid profiles (10, 115) ; these defects elicited by the Cav-1 knockout thus represent Cav-1 -specific effects. The pulmonary defects observed in both Cav-1 −/− and Cav-2 −/− null mice are most likely the result of a Cav-2 deficiency and are clearly independent of caveolae formation.
Caveolins and Pulmonary Disease
A single layer of capillary endothelial cells form a continuous lining and represent around 30% of the total cells in the alveoli (141, 142). The surface of these thin (<1 μ m) septa are enriched in caveolae, as pneumocytes express high levels of both Cav-1 and Cav-2 (143). The density of caveolae within this tissue suggests an important functional role in lung physiology, yet insight into their significance is surprisingly scarce. Indeed, a list of proposed caveolar functions pervades the literature, including a role in gas exchange, signal transduction, and endocytosis (143).
The lungs of both Cav-1 −/− and Cav-2 −/− mice have constricted alveolar spaces, associated with a hypercellular thickening of the alveolar walls and hypertrophy of certain cells (i.e, type II pneumocytes). The alveolar wall, which is normally organized in double layers, is disorganized and multilayered in Cav-1 −/− and Cav-2 −/− mice (114, 115) ; the cell proliferation marker Ki67 is upregulated, as is the receptor tyrosine kinase Flk-1 (VEGF-R), a marker of nondifferentiated endothelial and hematopoietic cells (109). Although these data suggest that endothelial cells are responsible for the alveolar thickening, the von Willebrand factor (vWF), a clotting factor that typifies differentiated endothelial cells, is not expressed. Intriguingly, phosphorylation of Cav-2 at Tyr19, a modification catalyzed by c-Src that results in localization of the protein to sites of focal adhesion, may establish a docking site for Src homology domain-2-(SH2)-containing proteins (144). It is thus attractive to speculate that Cav-2 may have a role in the local proliferation of angioblastic cells that reside in the alveolar wall.
Lung tissue alone shows the striking hypercellularity that results from obliteration of Cav-1 or Cav-2 in vivo (109, 114, 115) ; however, excessive proliferation of Cav-1 −/− MEFs occurs in vitro and is characterized by an increase in the number of S-phase cells (109). Associated with the increases in endothelial cell proliferation are an increase in extracellular collagen deposition associated with fibrosis (114, 115) and a thickening of the basement membrane (109). Fibrotic pulmonary disorders often result from injury to the lung parenchyma and are characterized by non-specific fibrosis due to infiltrating cells and the deposition of extracellular matrix (145). One might thus speculate that the loss of Cav-2 disrupts the alveolar architecture that results in tissue derangement such as is seen in fibrotic pulmonary disorders. Both Cav-1 −/− and Cav-2 −/−mice are exercise-intolerant in swimming tests, and these data are consistent with the presence of a restrictive lung disease phenotype (109, 114, 115).
Cav-3 and Striated Muscle
The expression of Cav-3 is largely restricted to cardiac and skeletal muscle cells. Cav-3 is upregulated, at the levels of transcription and translation, during the differentiation of skeletal myoblasts in vitro (146). Cav-3 is localized to the sarcolemma and is transiently associated with the transverse membrane pockets (i.e., the T-tubule network) that penetrate into the muscle fibers during development (147) and allow for rapid rises in cytosolic calcium following membrane depolarization (148). Antisense inhibition of Cav-3 expression in cultured skeletal myoblasts precludes myoblast fusion and myotube formation (55), normal processess of skeletal muscle development. Like Cav-1, Cav-3 is essential for the biogenesis of caveolae and the modulation of signaling molecules in cardiac and skeletal myocytes (116, 149).
Cav-3 and Muscular Dystrophy
Cav-3 has been shown to associate with dystrophin, an important member of the dystrophin-glycoprotein complex (DGC) that spans the muscle cell plasma membrane and links the cortical cytoskeleton with the extracellular matrix (13, 146). Dystrophin is the product of the DMD gene, the X-linked recessive gene associated with Duchenne muscular dystrophy (DMD) (150). DMD afflicts young children and results in morbidity by early adulthood from respiratory failure (151). In the muscles of patients who have DMD, the Cav-3 protein is upregulated and there is an associated increase in the number and size of caveolae at the sarcolemma (152, 153). In the mdx mouse, a dystrophin-deficient model of DMD, these two characteristics are similarly reiterated in skeletal muscle (154). Furthermore, Cav-3 has been shown to interact with the C-terminal PPXY domain of β -dystroglycan, an integral membrane component of the DGC. This interaction is mediated by a WW-like domain in Cav-3 that can competitively inhibit dystrophin binding and can consequently alter the recruitment of dystrophin to the sarcolemma (155).
Transgenic mice that broadly overexpress Cav-3 show specific skeletal muscle defects: 1) an increase in the number of muscle cell caveolae; 2) an abundance of necrotic, hypertrophic, and immature skeletal muscle fibers; 3) the downregulation of dystrophin and β -dystroglycan proteins; and 4) elevated serum creatine kinase levels, consistent with muscle necrosis (156). Although these findings initially linked CAV-3 with the pathogenesis of muscular dystrophy, a direct confirmation of this association only came when two distinct mutations in the CAV-3 gene were identified in an autosomal dominant form of limb-girdle muscular dystrophy (i.e., LGMD-1sC) (29, 30). Specifically, LGMD-1C is linked to two mutations in the CAV-3 gene: the first is a missense mutation in the membrane-spanning region, and the second is a 9-base pair deletion that removes three residues in the scaffolding domain. Both mutations result in a severe reduction of the Cav-3 protein in muscle and a loss of caveolae at the sarcolemma (30, 149) ; the mutant protein acts in a dominant-negative manner by forming aggregates with the wild-type Cav-3 protein. The aggregates are retained in the endoplasmic reticulum (ER) and Golgi apparatus and are subsequently degraded by the proteasome (157, 158). Patients with LGMD-1C also manifest mislocalization of dysferlin, a muscle membrane protein, recently shown to interact with Cav-3 (159), the deficiency of which is well-established in Miyoshi myopathy and LGMD-2B. Elevated serum creatine kinase (i.e., hyperCKemia) is also typical of LGMD-1C (149) and has been linked to CAV-3 mutations; idiopathic hyperCKemia may thus be indicative of a Cav-3 deficiency in skeletal muscle (160, 161).
Hereditary rippling muscle disease (RMD) is an autosomal dominant human disorder characterized by mechanically triggered contractions that spread to neighboring fibers and cause visible ‘ripples’ to move over the muscle (162). RMD has been linked to a locus on 3p25 that spans the CAV-3 gene, and several CAV-3 missense mutations have been identified from patients with RMD (163). Mutations of CAV-3 underlie the allelism identified in dystrophic (i.e., LGMD-1C) and nondystrophic (i.e., hyperCKemia and RMD) muscle pathologies. Interestingly, a naturally occurring Cav-3 mutant protein (i.e., C71W) results in localization to the plasma membrane, association with caveolae, and inhibition of H-Ras signaling. The P104L mutant Cav-3 protein, on the other hand, is retained in the Golgi apparatus and has no effect on H-Ras signaling (164), and transgenic mice overexpressing the P104L mutant Cav-3 protein develop severe myopathy. There is a significant increase in nNOS activity in the skeletal muscle of these mice, suggesting a role for NO in the degeneration of Cav-3–deficient muscle fibers (165). The molecular aberrations that result in these muscular dystrophies may lead to new diagnostic tests, more accurate prognoses, and improved therapeutic strategies.
Cav-3 Knockout Mice
Degeneration of Skeletal Muscle and Transverse Tubule Abnormalities
Cav-3 −/−mice lack muscle cell caveolae, but express normal amounts of Cav-1 and Cav-2 and show normal caveolae in other tissues (116) ; however, the Cav-3 knockout elicits a number of myopathic changes consistent with moderate muscular dystrophy. Soleus muscle degenerates in the knockout animals by eight weeks of age, as does the diaphragm at eight to thirty weeks, but there is otherwise no effect on growth and motor movement relative to wild-type mice (166). Cav-3 +/– hemizygotes have no muscle myopathy, indicating an autosomal recessive transmission of the myopathic phenotype, which contrasts with the dominant-negative Cav-3 missense mutations associated with LGMD-1C (166). Skeletal muscle cells from Cav-3 −/− mice exclude the DGC from lipid raft domains and have a disorganized T-tubule network (116).
Cardiomyopathy
Cav-3 −/− mice develop cardiomyopathy characterized by cardiac hypertrophy, dilation, and reduced fractional shortening by four months of age (167) ; cardiomyocytes are hypertrophic and show exclusion of the DGC from lipid raft domains. Histologically, the cardiac muscle shows increased cellular infiltration with accompanying perivascular fibrosis. Mechanistically, there is hyperactivation of the Ras-p42/44 MAPK cascade, consistent with the known role of MAPK activation in cardiac myocyte hypertrophy, and the role of Cav-3 as a negative regulator of the Ras-p42/44 MAPK cascade (167).
Cav-1/Cav-3 Double-Knockout Mice
Severe Cardiomyopathy
Cav-1 /Cav-3 double knockout (dKO) mice have been generated by breeding Cav-1−/− with Cav-3−/−mice. The dKO animals completely lack morphologically identifiable caveolae. The mice are viable and fertile, but are deficient in all three caveolin proteins, because Cav-2 is degraded in the absence of Cav-1, and are afflicted with severe cardiomyopathy. By two months of age, there is a significant increase in the thickness of the left ventricle wall (Figure 6⇓). There is an associated dilation and hypertrophy of the left ventricle; increased levels of atrial natriuretic factor mRNA, a marker of cardiac hypertrophy, are also present. Microscopically, the cardiac muscle is disorganized with signs of inflammation and perivascular fibrosis. The cardiac myocytes also show severe signs of hypertrophy and degeneration (168).
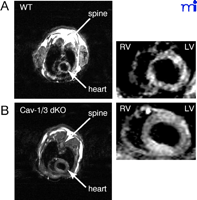
Effect of caveolin deficiency on the heart.Cav-1/Cav-3 double-knockout mice exhibit cardiac hypertrophy, as assessed by gated cardiac magnetic resonance imaging. Representative short axis (transverse) images at the mid-level of the heart of wild-type (A) and double-knockout (B) animals are shown during diastole. Note the concentric hypertrophy of the left ventricle (LV) in the caveolin-deficient heart. (RV, right ventricle.)
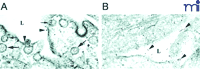
Caveolar endocytosis of BSA-conjugated gold is blocked in Cav-1-deficient lung endothelial cells. In the wild-type sample (A), caveolae open to the lumen (L) of the blood vessel are indicated by small arrows; large arrows indicate internalized caveolae. Arrow heads indicate gold particles in the lumen. In the Cav-1-deficient sample (B), caveolae do not form and gold particles therefore remain in the lumen.
Conclusions
The cell membrane is organized into discrete functional units, termed microdomains, that coordinate normal cellular activity. Caveolae microdomains contain caveolin proteins, and this molecular signature distinguishes them from other membrane domains. Nevertheless, it is likely that caveolae share common molecules and subserve functions overlapping with other microdomains. The composition, localization, and intracellular targeting of these caveolar microdomains are undoubtedly both tissue-specific and subject to regulatory control.
Not surprisingly, Cav knockout mice develop severe pathologies in lung, adipose, skeletal muscle, and other tissues that are morphologically rich in caveolae. Indeed, these knockout animals established definitive roles for Cav-1 and Cav-3 in the tissue-specific biogenesis of caveolae. In direct support of the idea that caveolae can regulate signal transduction [the “caveolae signaling hypothesis” (81) ], eNOS activity in the vasculature of Cav-1 −/− mice is significantly elevated. Unexpectedly, lung abnormalities result from Cav-2 ablation that are independent of caveolae formation. Additionally, there is severe cardiac hypertrophy in both Cav-1 −/− and Cav-3 −/− mice, a phenotype further exacerbated in Cav-1 /Cav-3 dKO mice.
Interestingly, Cav-1 −/− mice show no changes in serum cholesterol and no increases in the incidence of tumor formation, despite the wealth of in vitro evidence to support the role of Cav-1 in cholesterol homeostasis and in tumor suppression. However, the susceptibility of the Cav-1 −/− genotype to carcinogens and tumorigenic stimuli dramatically illustrates the important role of Cav-1 as a negative regulator of cellular proliferation and transformation. Future studies will be necessary not only to dissect the molecular pathways through which Cav-1 regulates cell growth, but also to determine whether the caveolins represent viable targets in therapeutic strategies to combat the pathologies seen in Cav knockout mice.
Acknowledgments
We thank Drs. Roberto Campos-Gonzalez (BD Pharmingen/Transduction Labs) for generously donating antibodies directed against Cav-1, Cav-2, and Cav-3. This work was supported by grants from the National Institutes of Health (NIH), the Muscular Dystrophy Association (MDA), the Susan G. Komen Breast Cancer Foundation, and the American Heart Association (AHA), as well as a Hirschl/Weil-Caulier Career Scientist Award (all to MPL).
- © American Society for Pharmacology and Experimental Theraputics 2003
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.↵
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.↵
- 167.↵
- 168.↵

Robert Hnasko, PhD (bottom), is a Postdoctoral Research Associate at The Albert Einstein College of Medicine. He is interested in the role of caveolae and caveolae-associated proteins in endothelial cell function. Address correspondence to MPL. E-mail lisanti{at}aecom.yu.edu; fax 718-430-8830.

Michael P. Lisanti, MD-PhD (top), is Professor of Molecular Pharmacology at The Albert Einstein College of Medicine. His laboratory focuses on elucidating the role of caveolae/caveolins in normal physiology and in the pathogenesis of human disease, with an emphasis on cancer and muscular dystrophy.

