Close Encounters of the Oily Kind: Regulation of Transporters by Lipids
Abstract
Neurotransmitter transporter function can be influenced by a variety of physiological factors, such as temperature, membrane voltage, pH, ion gradients, endogenous ligands, and accessory proteins. Several successful drugs that target transporter proteins, such as the selective reuptake inhibitors, have also been developed. One mechanism for regulating membrane protein function that has only started to make the list has been the impact of lipid environment. Over the last few decades, lipids, the unsung heroes of regulation have begun to emerge as critical modulators of membrane protein function through their direct actions on proteins, through their influence on the lipid milieu and through their roles in signaling pathways.
Introduction
Neurotransmitter transporters are essential to the homeostatic regulation of synaptic signaling throughout the nervous system. The extensive amount of research that has been devoted over the past few decades to these integral membrane proteins is an apt reflection of this importance. Transport mechanisms have been a prime target of the pharmaceutical industry, prompting the investment of billions of dollars into the development of drugs, such as the selective reuptake inhibitors, several of which have achieved blockbuster status in the treatment of depression and anxiety. The functions of many carriers are influenced by a variety of factors such as temperature, membrane voltage, pH, ion gradients, ligands, and accessory proteins. One mechanism for regulating membrane protein function that has only started to make the list has been the impact of lipid environment. Over the last few decades, lipids, the unsung heroes of regulation, have begun to emerge as critical modulators of membrane protein function through their direct actions on proteins, through their influence on the lipid milieu, and through their roles in signaling pathways.
A variety of mechanisms have been identified through which transporters act to regulate the storage, subcellular localization, and synaptic availability of neurotransmitters. Physiological factors that influence transporter function include temperature, membrane voltage, pH, ion gradients, endogenous ligands, and accessory proteins. The degree to which any particular mechanism contributes to transporter function or might be exploited for improved drug development is not always clear. But the importance of transporters in basic neurobiology and translational medicine certainly mandates investigation from a variety of approaches. The advent of lipidomics in past years has enriched our understanding of signal transduction in many contexts, including neurotransmission. The versatility of lipids in signaling has proven remarkable, and one aspect of this signaling is the role of lipids as modulators of regulatory proteins in the cell membrane.
Lipids, once viewed simply as building blocks of the membrane, have for many years now been appreciated for their role in signal transduction at the cell surface. Receptor- and enzyme-mediated hydrolysis of PIP2, producing DAG and IP3, has become a canonical feature of signaling cascades. Fatty acid amides and arachidonate metabolic products, including a fascinating array of endocannabinoids, are involved in regulating physiological and basic behavioral states through the activation of specific cell surface receptors. [For an excellent review of the contributions of lipids to cellular signaling, see (1).] The direct binding and allosteric modulation of proteins by lipid molecules has been proposed in many systems, yet additional subtle effects of lipids on protein function can also occur through variations in the structure, rigidity, or lateral pressure of the lipid components affecting protein conformation or dynamics.
This review will emphasize the direct effects of membrane lipids on transport proteins relevant to the central nervous system. [For a review of the actions of lipids on ion channel properties, see (2–4)]. We will attempt to complement traditional concepts of the neural cell membrane with data focusing on the interaction of both cholesterol and fatty acids with plasma membrane carriers. We will provide common names to discuss particular lipids (see Table 1), but we will also rely on the LIPID MAPS initiative, with its more precise nomenclature and comprehensive systematization of lipids and their metabolism (5). In accordance with much of the literature, we occasionally refer to “simple” lipids as those that produce only one or two products upon hydrolysis, as opposed to “complex” lipids, which yield a larger number of hydrolysis products.
Eight Lipid Catagories and Subclassification According to LIPID MAPSa
Transporter Regulation by Polyunsaturated Fatty Acidsa
Transporter Responses to Alteration of Membrane Cholesterol Contenta
Lipid Composition of Cellular Membranes
Many initial efforts to characterize the “lipidome” of neural tissues were limited in resolution to only a few of the predominant membrane constituents. For example, lateral vestibular neurons were reported to contain cholesterol [ST01010001], phosphatidylethanolamine [GP02], phosphatidylserine [GP03], oleic acid [FA01030002], stearic acid [FA01010018], palmitic acid [FA01010001], as well as phosphoinositols [GP06] (6). Differences in lipid composition between axonal plasma membranes and the surrounding myelin were in a sense some of the earliest “lipidomics” data: axonal membrane was reported as 20.1 % cholesterol, 20.1 % galactolipid [SP06], 14.6% phosphatidylethanolamine, 18.3% phosphatidylcholine [GP01], 9.3% sphingomyelin [SP0301], 5.6% phosphatidylserine, and 3.4% phosphatidylinositol [GP06]). Ceramides [SP02] and sulfoglycosphingolipids [SP0602] predominated as the 24:1 unsaturated form. In myelin, the saturated version of sphingolipid (i.e., 24:0) predominated (7–9). From this early work, it became clear that the plasma membrane of neurons (and indeed of most mammalian cells) is comprised of a combination of cholesterol, phosphatidyle-thanolamine, phosphatidylserine, phosphatidylcholine, sphingo-myelin, and glycolipids in an asymmetrical leaflet distribution. [For a comprehensive review of history of early neuronal lipidomics, see (10).]
To understand how the lipid environment influences protein function it is imperative to define not only the lipid components of the membrane, but their organization in the membrane. It was shown almost fifty years ago that artificial lipid monolayers can transition from a gel-like state to a liquid-crystalline state with increasing temperature (11). In the gel phase, acyl chains in a predominately all-trans configuration undergo orderly packing, whereas in the liquid crystalline state acyl chains with disordered or random orientations predominate (Figure 1). Lipid bilayers can also transition between these states, and the plasma membrane is best modeled as a liquid-disordered state (12). Although the study of lipid monolayers and pure or controlled lipid environments has revealed much about dynamics of lipids in general, such studies cannot offer a comprehensive model of a biological membrane that contains integral membrane proteins. In 1972, Singer described a model of the plasma membrane as a two-dimensional dynamic viscous solution in which integral membrane proteins maximize their hydrophobic and hydrophilic interactions in accordance with the surrounding lipid matrix and aqueous environment (13). Singer’s “fluid mosaic model” has been well supported for over thirty years (14), and an extensive literature reveals that variability in lipid composition affects viscosity, depth, curvature, and lateral pressure (15–19).
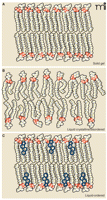
The dynamic lipid bilayer and membrane phases. Three different phases are generally described for membrane lipids. A) In the “solid gel state,” or “solid crystalline state,” hydrocarbon chains are typically found in the trans configuration and are highly ordered. Low temperatures can favor the solid gel phase. B) This “liquid crystalline state,” or “liquid disordered state,” is typical of high temperature and shows a disordered configuration of acyl chains. High lateral mobility is associated with the liquid disordered state; most pure fatty acid, detergent, or phospholipid liposomes are found in this state at room temperature. C) The “liquid ordered state” occurs in the presence of high levels of sphingolipids and cholesterol and results in mobility slightly under that of the pure liquid disorderd state. Detergent-resistant microdomains or lipid rafts are endogenous liquid-ordered membrane patches. See text for details. Adapted from (63).
Singer also recognized, in certain membrane regions, the possibility of liquid-ordered phases, characterized by restriction in the lateral movement of lipids. The emergence of lipid rafts as a field of study, although not without its share of debate, has established that local lipid composition can have profound impacts on integral protein structure and activity. Lipid rafts, or detergent resistant microdomains (DRMs), are defined as cholesterol- and sphingo-myelin-rich membrane regions that impose an ordered state to the surrounding lipid environment (20, 21). Although not considered in detail here, this area of research has brought an exciting new focus of research and discussion that will further our understanding of lipids and membrane dynamics and how they relate to biological function.
Influence of Fatty Acids on Transporter Function
Polyunsaturated fatty acids (PUFAs; [FA0103]), such as arachidonic acid [FA01030001], comprise twenty to thirty percent of free fatty acids in the human brain (22). Typically derived from diet, polyunsaturated fatty acids in the brain are either transported from plasma and esterified into complex lipids or are released from membrane glycerophospholipids (23). Levels of brain PUFAs can increase with neuronal activity (e.g. activation of NMDA-type glutamate receptors) and area subsequently metabolized by lipases, typically phospholipase A2 (PLA2) (24). Arachidonic acid (and other PUFAs) can modulate both the electrical and biochemical properties of a variety of membrane proteins involved in cellular signaling mechanisms, including ion channels, receptors, and transporters. A critical question in all these examples is whether arachidonic acid modulates protein activity through a direct interaction with the protein or whether it indirectly modulates the protein either by altering membrane fluidity or through the actions of metabolites (e.g., mediated by cyclooxygense and lipoxygenase pathways).
The effects of arachidonic acid on brain transporter activity were initially examined in assays of glutamate uptake, γ-aminobutyric acid (GABA) uptake, and Na+/K+-ATPase activity (25). Incubation of rat cortical slices with arachidonic acid caused a dose-dependent reduction in the Na+-dependent transport of GABA and glutamate but did not affect transport of α-aminoisobutyric acid. Na+/K+-ATPase activity was reduced not only by arachidonic acid but also by docosahexaenoic acid [FA01030185]; the basis for these responses did not appear to stem from any detergent-like actions, as the addition of sodium dodecylsulfate at low concentrations had no effect. Similarly, in rat primary neuronal cultures, uptake of both glutamate and GABA was significantly inhibited by the two PUFAs, whereas glutamate transport alone was altered in astrocytes (26). The saturated fatty acid palmitate had no effect on transport in either neurons or glial cells. Taken together, these studies suggested that PUFAs modulate transport systems in a cell-type specific manner, perhaps related to intrinsic differences in membrane composition. activator, has been used to
Melittin, a phospholipase A2 increase cellular production of arachidonic acid from endogenous glycerophospholipid precursors. In one study, the addition of melittin together with an acyltransferase inhibitor to cultured astrocytes increased arachidonic acid levels in a dose-dependent manner and concomitantly inhibited glutamate uptake (27). This inhibition is insensitive to the cyclooxygenase inhibitor indomethacin but is abolished upon application of bovine serum albumin, which binds free fatty acids. These experiments were not able to determine whether arachidonic acid affects the glutamate transporter through a direct interaction with the protein or whether it inhibits glutamate transport through an indirect mechanism. The metabolism of arachidonic acid generates free radicals and reactive oxygen species (ROS), which are reported to inhibit glutamate transport in cells; thus, ROS were examined as potential indirect means for arachidonic acid–mediated inhibition of glutamate transport (28–31). In astrocyte cultures treated with an ROS-producing agent, saturating doses of arachidonic acid produced further inhibition of glutamate uptake. In addition, transport inhibition was fully prevented in cultures treated with both BSA (to sequester arachidonic acid) and superoxide dismutase/catalase (to prevent the accumulation of ROS). Application of either agent alone recovered only a portion of the inhibition, confirming that arachidonic acid mediates transport in an ROS-independent manner. These investigations elegantly demonstrated that PUFA modulation is a distinct mechanism and not dependent on oxidative stress or related processes.
In isolated salamander retinal glial cells, arachidonic acid can inhibit the glutamate-evoked currents that arise from electrogenic glutamate uptake. Whole cell patch recordings of salamander Muller cells reveal that arachidonic, docosahexaenoic, and oleic acids reduce maximal transport currents without dramatically affecting glutamate affinity (32). These inhibitory effects did not appear to depend on metabolic turnover of the PUFAs, as the inhibition was affected by neither nordihydroguaiaretic acid nor indomethacin, inhibitors of lipoxegenase and cycooxygenase, respectively. Using a different model system, Trotti and colleagues investigated the effects of a series of fatty acids on the uptake of radiolabeled glutamate in reconstituted proteoliposomes enriched in the glutamate transporter isoforms Glt-1 (33). They showed that oleic acid, methyl ester-arachidonic acid, α-linolenic acid [FA01030152], docosahexaeonic acid, and eicosapentaenoic acid [FA01030181] all inhibited glutamate transport, and interestingly, eicosapentaenoic acid inhibited uptake significantly more than did arachidonic acid. A major finding from this work was that arachidonic acid present in the aqueous but not the lipid phase was essential for the inhibitory effect. This observation supports the intriguing possibility that the inhibition of glutamate transport is the consequence of a direct interaction between arachidonic acid from the aqueous medium and the transporter protein. Remarkably, in 2004, the crystal structure of an archaeal ortholog of mammalian glutamate transporters revealed the presence of a lipid moiety bound within the solvent-filled basin. In this structure the lipid extends down into a hydrophobic crevice that is observed only in the substrate-bound carrier (34) (Figure 2A). This state-dependent lipid interaction at the aqueous interface of a glutamate transporter is intriguing and may provide a structural link to the lipid-mediated inhibition of carrier activity noted above.
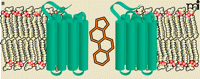
Lipid moieties found within high-resolution structures of integral membrane proteins. A) A high-affinity aspartate transporter (GltTph) from Pyrococcus hirokoshii resolved at 3.2Å. Surface representation of a single GltTph monomer, colored by atom type, with the interacting palmitate molecule colored green. At right, the hydrophobic residues that are positioned to interact with the lipid molecule are shown. B) Membrane topology of the human β2-adrenergic receptor based on the crystal structure resolved at 2.4 Å. Intriguingly, association of the two receptor molecules (blue) appears to be mediated by cholesterol (orange). [Adapted from (34, 64).]
The question of whether PUFAs act directly or indirectly to modulate transporter function was further addressed in investigations of the effects of arachidonic acid and other fatty acids on transport currents associated with human excitatory amino acid transporters (EAATs) (35). Because transport of glutamate by EAATs is coupled thermodynamically to the co-transport of one proton and two to three sodium ions, and to the counter-transport of one potassium ion, the process is electrogenic and can be studied using electrophysiological techniques in Xenopus oocytes. With all EAAT subtypes, most notably EAAT4 and EAAT5, substrates can additionally activate a thermodynamically uncoupled anion flux. Three glutamate transporter isoforms were expressed in Xenopus oocytes and tested for their sensitivity to arachidonic acid: EAAT1, a glial carrier; EAAT2, an abundant, predominantly astrocytic carrier; and EAAT3, a widely expressed neuronal carrier. The effects of PUFAs were distinct for each transporter subtype: maximal transport activity of EAAT 1 was decreased without alteration of glutamate affinity; the activity of EAAT2 was increased by decreasing its apparent Km for glutamate; and little or no effect was observed on the activity of EAAT3. These results, together with synaptosomal studies reporting different arachidonic acid effects on glutamate transport in various regions of the rat central nervous system (36), first illustrated the subtype-selective actions of arachidonic acid on glutamate transporters. Proteoliposomal preparations further corroborate these observations, raising again the possibility that PUFAs such as arachidonic acid regulate transport through direct interactions with the carriers themselves, rather than through indirect effects on membrane organization or fluidity. Moreover, the rapid onset of modulation, along with its insensitivity to cyclooxygenase and lipoxygenase inhibitors, suggests that the effects on glutamate transport are mediated directly by arachidonic acid itself, rather than via a metabolite.
Further insight into the ability of arachidonic acid to differentially modulate the activity of glutamate transporters comes from investigation of human EAAT4, a carrier that possesses a prominent substrate-activated anion current (37). Although arachidonic acid does not significantly affect substrate transport, it causes a large increase in substrate-activated EAAT4-mediated currents. Ion substitution experiments have revealed that EAAT4 generates a distinctive substrate-gated proton-selective current that is PUFA-dependent. The proton conductance, activated at low, physiologically relevant, concentrations of arachidonic acid (EC50 ≈ 2μM), is not associated with changes in substrate flux, PUFA metabolism, ion coupling, or the well-established anion conductance. The phenomenon is characteristic of rat as well as human EAAT4 (38). Thus, several lines of data indicate that arachidonic acid and other PUFAs alter the channel-like signaling properties of the EAAT4. [For reviews of EAAT modulation by PUFAs, see (39, 40)].
In addition to the excitatory amino acid family of carriers, which belong to solute carrier family 1 (SLC1), a second family of transporters, the biogenic amine carriers, are also regulated by PUFAs. These carriers are members of the neurotransmitter–sodium symporter family (SLC6). For example, arachidonic acid inhibits the uptake of L-dopamine into rat striatal synaptosomes in a dose-dependent manner (41). In contrast, C6 glioma cells stably expressing the dopamine transporter respond to arachidonic acid (20–160μM) in a biphasic manner: dopamine transport is activated in the short term, but at later time points (incubations > 45 min), transport is inhibited relative to control (42). Thus, within a single cell line, arachidonic acid may have multiple modes of action on the dopamine transporter.
In addition to the electrogenic co-transport of Na+, Cl− and dopamine, the dopamine transporter mediates a sodium-dependent cation leak conductance (43), which is relevant in determining the specific effects of PUFAs upon the transporter. Indeed, like the excitatory amino acid transporter discussed above (EAAT4), the dopamine transporter responds to arachidonic acid by displaying unique conductances. The application of arachidonic acid (1–100 μM) to Xenopus oocytes expressing the human dopamine transporter stimulates an inward current that is potentiated by dopamine and can be blocked by transport inhibitors such as cocaine (44). The current is qualitatively and quantitatively different from the currents typically associated with dopamine transport in several ways: it shows a distinctive selectivity for cations; its amplitude can be up to fiftyfold larger than current arising from electrogenic transport; and it is more pH-sensitive than transport. In voltage-clamped oocytes, arachidonic acid inhibits dopamine transport. Interestingly, dopamine, even in the absence of its co-substrate sodium, can potentiate the arachidonic acid–induced current. These effects appear to be produced by a direct action of arachidonic acid on the carrier, as they are not mimicked by non-metabolizable analogs and they are insensitive to a variety of inhibitors of fatty acid metabolism.
Because many studies have ruled out changes in fluidity or other membrane dynamics as contributors to the effect of PUFA actions on transporters, the observation that a range of these lipid molecules can mediate similar effects may well reflect a generally accommodating binding site. Nonselective binding is unlikely to be as energetically favorable as the binding of ligand to a more specific binding site. Nevertheless, the presence of lipid moieties within a number of high-resolution membrane protein structures lends additional credence to the possibility of allosteric lipid modulation (Figure 2).
Cholesterol and Transporter Function
Cholesterol is another constituent of the membrane that has been shown to have a major effect on protein dynamics and function. Increases in membrane cholesterol content have been shown to affect lipid fluidity and to increase the viscosity of the membrane (45). The Na+/K+-ATPase is one of the most extensively studied carriers in biology, and early studies indicated that membrane cholesterol depletion increases pump activity. To establish whether cholesterol depletion alters maximal transport rates through a general change in membrane lipid fluidity or through a more direct mechanism, the effect of several additional amphiphilic molecules that enhance membrane fluidity were tested (46). Electronic spin resonance and fluorescence polarization measurements with these compounds revealed a membrane-disordering effect that mimicked cholesterol removal. All treatments decreased the apparent internal Na+ affinity but increased maximal transport velocity of the ATPase. In addition, the supplementation of bovine kidney membrane vesicles with phosphatidylcholine and sterols evoked a biphasic response in pump activity, in which low levels of cholesterol stimulated activity, whereas high concentrations inhibited ATP hydrolysis (47).
Although initial studies consistently demonstrate a biphasic action of cholesterol on ATPase activity, the magnitude of inhibition observed varied widely among the various artificial membrane and tissue preparations studied. A comprehensive series of experiments addressed the effects of cholesterol on the kinetic properties of the Na+/K+-ATPase isolated from the shark rectal gland; pump activity was reconstituted in liposomes consisting of phosphatidylcholine, phosphatidylinositol, and phosphatidylethanolamine. The addition of cholesterol to these liposomes caused a significant increase in steady-state rate of Na+/K+-exchange activity, Na+/Na+-exchange, and on Na+ uncoupled transport (48). Additionally, cholesterol increased the rates of ATPase phosphorylation and dephosphorylation by as much as forty percent. These data supported a model in which the incorporation of cholesterol into liposomes shifts the equilibrium of ATPase conformers, accounting for kinetic effects observed for the potassium deocclusion reaction as well as for the observed change in phosphoenzyme levels (49).
The fatty acid composition of the lipid bilayer can also affect Na+/K+-ATPase activity. Unsaturated fatty acids, which display fewer packing interactions and higher fluidity than saturated fatty acids, give rise to higher levels of ATPase activity in proteoliposome preparations (50). Additionally, acyl chain length is a variable that has been demonstrated to impact protein activity with optimal functioning of the ATPase in lipid vesicles containing between 16- and 20-carbon atom phosphatidylcholine acyl chains (51). When the Na+/K+-ATPase was assessed in a reconstituted liposome system in the absence of cholesterol, the optimal acyl chain length for a monounsaturated phospholipid was twenty-two carbons, whereas the optimal length in the presence of cholesterol was shifted to 18 carbons (52). No significant effect on pump activity was seen in response to temperature alteration supporting the notion that that bilayer thickness is a more critical variable for regulating activity than membrane fluidity. Subsequent work also revealed that interactions between cholesterol and the polyunsaturated acyl chains of lipids can significantly influence Na+/K+-ATPase activity. Specifically, the addition of phosphatidylcholine molecules containing docosahexaenoic acid acyl groups lowers pump activity and decreases Na+ binding (53), effects that vary inversely with the amount of cholesterol incorporated into the membrane. The docosahexaenoic acid moiety, with its six double bonds, induces a high degree of disorder within the membrane, whereas cholesterol favors a liquid-ordered state. Docosahexaenoic acid-containing phospholipids thus tend to segregate laterally away from cholesterol and establish liquid-disordered domains. These experiments demonstrate a fundamental effect of membrane properties on integral protein function, and also reveal more specific interactions of cholesterol and phospholipids with the ATPase.
Clearly, studies of Na+/K+-ATPase activity have established that the activity of an integral membrane protein can be regulated by membrane lipid composition, bilayer thickness, and general fluidity. Membrane cholesterol has also been shown to modulate the activity of neurotransmitter transporters. Transport by a rat brain GABA transporter reconstituted into liposomes was significantly impaired when the lipid bilayer consisted solely of asolectin (soybean phospholipids), but transport activity was restored upon addition of brain lipids containing cholesterol (54). This stimulation was specific in that it could not be replicated by cholesterol analogs. Brain glutamate transport in proteoliposomes is similarly cholesterol-dependent, and the stimulatory effect of cholesterol in this case could not be explained by any changes in ion coupling or carrier surface expression. Remarkably, very modest amounts of cholesterol are required to restore GABA or glutamate transport activity, implying that a direct interaction of cholesterol with the carriers may mediate this effect. A more recent study has explored a distinct effect of cholesterol in primary neuronal cultures from rat cortex. The intrinsic glutamate transport activity of pure neuronal cultures is lower than that of neurons co-cultured with astrocytes (55). The transport deficiency manifested by pure neuronal cultures can be rescued by astrocyte-conditioned media, and cholesterol itself was identified as the molecule responsible for the observed increase in transport function. One additional study of the effect of cholesterol depletion on glutamate transport in rat cortical cultures showed that when the sterol chelator methyl-β-cyclodextrin (MβCD) was used to remove cholesterol, radiolabeled glutamate uptake was significantly reduced. Additional experiments showed that EAAT2 resides in detergent-resistant lipid microdomains and that MβCD treatment altered this localization (56).
Membrane cholesterol content can also regulate the activity of the serotonin transporter, which also belongs to the Neurotransmitter–Sodium Symporter family (NSS or SLC6 family). Treatment of HEK293 cells stably expressing the rat serotonin transporter with MβCD decreases maximal transport rate, lowers the apparent affinity for serotonin, and abolishes binding of the antagonist citalopram (57). These effects were specific for cholesterol and not due to alterations in membrane fluidity, because analogs of cholesterol that alter membrane fluidity had no effect on transport activity. In addition, disruption of lipid microdomains upon removal of cholesterol with MβCD has been linked to reduced SERT activity (58).
The human dopamine transporter is also regulated by its association with membrane lipid microdomains and by changes in cholesterol content (59). Based on fluorescence recovery after photobleaching (FRAP) experiments, an association of the dopamine transporter (tagged with yellow fluorescent protein) with lipid microdomains was reported in HEK293 cells. With the depletion of membrane cholesterol by MβCD treatment, the lateral mobility the dopamine transporter was increased, and this effect was associated with a decline in the transporter Vmax as well as its apparent affinity for dopamine. Interestingly, the disruption of the cytoskeleton by cytochalasin D also disrupted the localization of the dopamine transporter in this system, although it was unclear whether this effect alone was sufficient to alter transporter activity. Another study of the dopamine transporter, in which the effect of protein kinase C on dopamine transport was examined with respect for the distribution of the transporter at lipid microdomains (60), concluded that 1) the regulation of the dopamine transporter by cholesterol occurred within lipid rafts; and 2) that cholesterol-dependent regulation of dopamine transport was not mediated through alterations in cell surface expression of the transporter. In addition, studies examining the accessibility of cysteine residues introduced into the carrier sequence have demonstrated an increase in the number of cocaine binding sites when cells are provided with medium enriched with water-soluble cholesterol (Hong and Amara, unpublished data). These results support the notion that membrane cholesterol affects the conformational equilibrium of the transporter domains to favor an outward-facing state.
Two additional members of the SLC6 family, the glycine transporters GLYT1 and GLYT2, regulate glycine concentrations near central nervous system synapses. The neuronal carrier, GLYT2, localizes to detergent-resistant microdomains that are required to maintain full activity, and both cholesterol removal and sphingomyelinase treatment reduce GLYT activity (61). In contrast, a second study, using human GLYT1 and GLYT2 stably expressed in Chinese hamster ovary cells, reported that MβCD-mediated depletion of cholesterol reduced GLYT1 activity but actually increased GLYT2 activity (62). The same experiments, repeated in Xenopus laevis oocytes, revealed that although GLYT1 catalytic activity was reduced, GLYT2 activity was unaltered. Differences among these observations could arise from cell-specific differences in membrane thickness, fluidity, composition, or the presence of specific regulators.
Conclusions
The precise mechanisms for regulation of both primary and secondary carriers by lipids have remained elusive, in part reflecting the diversity of pathways and settings in which lipids can alter the structure and function of membrane proteins. Emerging data on the functional impact of lipids on membrane proteins, together with new high-resolution structures of protein–lipid complexes, support the idea that direct interactions of lipids and proteins will be found in many different transport systems. Nonetheless, the indirect effects of fatty acid composition, membrane fluidity, lateral segregation of membrane components, and bilayer dimensions in complex membrane environments will undoubtedly be part of the picture, too. Despite all the possibilities, the intimate association of membrane proteins with the lipid milieu guarantees that their functions will be shaped and regulated by a diverse, but essential cadre of oily characters.

- Copyright © 2009
References

Susan G. Amara, PhD, (right) is the Thomas Detre Professor and Chair of the Department of Neurobiology at the University of Pittsburgh School of Medicine and Co-director of the Center for Neuroscience at the University of Pittsburgh. Work in her laboratory focuses on neurotransmitter transporters and the plasma membrane proteins that limit the actions of neurotransmitters following their release from neurons. Christopher B. Divito, BA, (left) graduated from Shippensburg University of Pennsylvania with concentrations in psychology and biochemistry. He is currently a third-year graduate student, earning his PhD at the Center for Neuroscience of the University of Pittsburgh. Chris’s research is focused on ligand binding mechanisms and lipid–protein interactions within the family of mammalian glutamate transporters.

