Strontium Ranelate in Osteoporosis and Beyond: Identifying Molecular Targets in Bone Cell Biology
Abstract
Osteoporosis is characterized by reduced bone mass and deterioration of bone microarchitecture, resulting in bone fragility and increased susceptibility to fractures. Current antiosteoporotic treatments depend on antiresorptive or anabolic drugs, but a novel modality of treatment appears to be mediated by strontium ranelate, which has been shown to act by opposing bone resorption and formation in vitro. This review article addresses the cellular and molecular mechanisms that have been implicated in the therapeutic strengthening of bone observed upon administration of strontium ranelate to osteoporotic patients. These mechanisms relate to specific pathways of calcium signaling, including complex networks involving nuclear factor of activated T cells (NFAT) and Wnt signaling.
Introduction
Maintenance of the adult human skeleton occurs through the same processes of bone remodeling that promote early bone development. The continual remodeling of bone matrix in the support of skeletal homeostasis is ensured by two cell types: the osteoclasts, which resorb calcified bone matrix, and the osteoblasts, which are responsible for the synthesis of new bone matrix. With aging, and particularly after menopause, an imbalance in bone resorption relative to bone formation can disrupt trabecular connections and microarchitecture, which is clinically manifested as reduced bone strength and in increased risk of fracture (1, 2).
Pharmacological compounds have been developed to combat osteoporosis by decreasing bone resorption (3, 4). Chief among these are the bisphosphonates, which potently inhibit bone remodeling activities (both resorption and formation) and have been proven effective at reducing the incidence of bone fractures associated with osteoporosis (5, 6). In contrast to the bisphosphonates, parathyroid hormone (PTH), when administered intermittently to osteoporotic patients, acts as an anabolic agent and favors bone formation relative to bone resorption, leading to increased trabecular bone mass and a reduction in fracture risk (7, 8). The strontium salt of renalic acid, a metal-chelating organic acid, also has antiosteoporotic activity, but its mode of action is distinct in that it promotes bone synthesis in addition to inhibiting resorption (9). Clinical trials have shown that strontium ranelate is effective in reducing fracture incidence in osteoporotic postmenopausal women (10, 11); however, despite important therapeutic effects in osteoporosis, the molecular mechanisms by which strontium ranelate acts to increase bone mass and strength are not fully understood.
This review provides an overview of the development of strontium ranelate as a therapeutic agent in osteoporosis. The discussion will extend to recent advances in the identification of molecular mechanisms of its action on bone cells and also highlights important unresolved issues regarding the mechanisms by which strontium ranelate may increase bone strength in osteoporotic patients.
Effects of Strontium Ranelate on Bone Cells
Strontium is a trace element that resides directly beneath calcium in the periodic table, and like calcium, strontium has a high affinity for bone (12). It has in fact been known for some time, however, that strontium administration in rodents increases vertebral bone volume, a result that is in contrast to calcium supplementation. This intriguing effect on bone mass results both from decreased bone resorption and increased bone formation, as assessed by histomorphometric analysis of rodents that had been administered strontium (13–16). These findings first raised the interesting possibility that strontium might be of potential benefit in the treatment of osteoporosis. Consistently, strontium ranelate, composed of two stable strontium atoms chelated by an organic acid, was found to induce opposing effects on bone resorption and formation in several experimental settings (17) (Table 1).
Effects of Strontium Ranelate on Bone Cells In Vivo.
Strontium ranelate was initially shown to reduce osteoclast activity and bone resorption in vitro (18). One of the cellular mechanism by which strontium ranelate may reduce osteoclast activity is through disruption of the cytoskeleton (19). In addition, strontium ranelate at high doses (25 mM) was shown to promote osteoclast apoptosis (20), thereby providing a basis for decreased bone resorption in vitro (20–22).
At the same time, strontium ranelate also induces positive effects on bone formation in vitro. Specifically, it enhances the replication of preosteoblastic cells, which supply the bone matrix with functional osteoblasts that drive bone synthesis (23–26). Strontium ranelate appears to be involved in osteoblast differentiation, as it facilitates osteogenesis and the expression of phenotypic markers in cultured osteoprogenitor cells or osteoblasts (21, 27–29). The net effects of strontium ranelate (i.e., reduction of osteoclast activity and promotion of osteogenic differentiation) thus distinguish the drug from the bisphosphonates (which act predominantly to inhibit bone remodeling) and intermittent PTH therapy (which acts to stimulate bone remodeling) (3).
Molecular Targets of Strontium in Bone Cells
It is apparent that strontium can act through multiple mechanisms to affect signaling pathways in bone cells.
Role of the Calcium-sensing Receptor
One important target of strontium is the extracellular calcium-sensing receptor (CaSR) (Figure 1). The CaSR is a heptahelical transmembrane-spanning G protein–coupled receptor (GPCR) that senses extracellular calcium. The CaSR plays a key role in maintaining extracellular calcium ion concentrations, mediating renal calcium reabsorption as well as the inhibitory effects of calcium ions on PTH secretion (30). The parathyroid CaSR homolog is expressed in cells of the osteoblast lineage (31–33). The CaSR is also expressed in osteoclast precursor cells and osteoclasts (34–36).
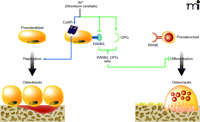
Mechanisms of action of strontium ranelate on bone cells. Activation (blue arrows) of the calcium sensing receptor (CaSR) by strontium ranelate in preosteoblasts (blue nucleus) increases osteoblast replication and number, which in turn results in increased bone formation. Strontium ranelate also acts on preosteoblasts (green arrows) to decrease the expression of RANKL (which binds RANK in preosteoclasts) and increases the expression of the decoy receptor osteoprotegerin (OPG), a dual effect that in turn results in reduced osteoclast differentiation and decreased bone resorption.
The importance of the CaSR in the regulation of bone cells has been established through both biochemical and genetic studies (37, 38). In multiple species, it promotes the chemotaxis and proliferation of osteoblasts (39–41). The CaSR is also involved in osteoblast differentiation, as high calcium levels promote the expression of osteoblastic differentiation markers and osteogenesis in cell culture (42); accordingly, abolition of CaSR function decreases osteoblast-specific gene expression (33). Thus, multiple lines of evidence indicate in both osteoblasts and osteoclasts that extracellular calcium concentrations may control cell proliferation and differentiation through activation of the CaSR (43, 44).
Extracellular calcium–activation of the CaSR invokes multiple cellular signaling pathways, including those dependent on phospholipase C (PLC), protein kinase C (PKC) (45), extracellular signal regulated kinases (ERK1/2) (40), Jun kinase (JNK) (41), and cAMP/protein kinase A (PKA) (46). Notably, the increased osteoblastic cell replication induced by calcium is mediated by CaSR-induced activation of ERK1/2 signaling (26, 41). Additionally, activation of CaSR activates the pro-survival Akt pathway that mediates calcium-dependent osteoblast survival (26). In osteoclasts, activation of the CaSR also triggers various signals that control gene expression and cell survival (47). Specifically, CaSR activation in vitro increases osteoclast apoptosis through activation of PLC and NF-κB (20). Moreover, CaSR-mediated activation of PI3K/Akt signaling stimulates the migration of osteoclast precursor cells (48). Thus, the CaSR appears to play important roles in the control of both osteoblastogenesis and osteoclastogenesis (49, 50).
Intriguingly, strontium can bind and activate CaSR as a full agonist, although with a lower affinity than calcium, and can thereby activate PLC and ERK1/2 (25, 51). Additionally, strontium can modulate intracellular calcium levels via inositol trisphosphate (InsP3) receptors and PLC activity (52, 53). In cultured osteo-clasts, strontium ranelate–induced activation of the CaSR results in caspase-mediated apoptosis (20) through mechanisms that differ from those induced by extracellular calcium. Indeed, extracellular calcium–induced activation of CaSR leads to IP3 activation NF-κB translocation, whereas strontium activates PLC, PKCβII, and NF-κB translocation (20). In osteoblasts, strontium-induced activation of the CaSR results in enhanced ERK1/2 signaling and increased osteoblastic cell replication (25, 26, 54). Strontium ranelate also increases the replication of cells of the osteoblastic lineage by activating PKC, PKD, and p38 MAPK pathways (24). Thus, strontium ranelate activation of the CaSR in bone cells engages multiple downstream signaling pathways that control cell proliferation, differentiation, and survival (Figure 2).
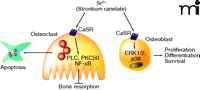
Signaling pathways induced upon activation of the calcium sensing receptor by strontium ranelate in bone cells. In osteoclasts, activation of the calcium sensing receptor (CaSR) by strontium ranelate induces activation of PLC, PKCβII, and NF-κB translocation, resulting in osteoclast apoptosis. In osteoblasts, strontium ranelate–induced activation of the CaSR induces activation of a number of kinases (e.g., ERK1/2 and p38MAPK, PLC, PKD, and PI3K), resulting in increased preosteoblast replication, osteoblast differentiation, and survival.
The effects of strontium on osteoblasts do not appear to rely solely on the CaSR, as engineered mice that lack the receptor can respond to strontium programmatic changes in gene transcription (55). Moreover, strontium ranelate can increase cell replication in primary osteoblast cultures from mice that lack the CaSR (26). Additionally, strontium ranelate can prevent the induction of apoptosis in cultured CaSR-null osteoblasts exposed either to serum-deficient medium or pro-inflammatory cytokines (26).
Other than the CaSR, cation-sensing receptors that have been implicated in the effects of strontium on osteoblasts include the orphan receptor GPRC6A, which is similar to the CaSR and can sense extracellular divalent cations (56). GPRC6A is expressed by osteoblasts and can subserve calcium ion sensing, and significantly, strontium activates ERK1/2 signaling through GPRC6A activation in vitro (57). Furthermore, GPRC6A-deficient mice develop osteopenia (i.e., low bone density) and show attenuated responsiveness to calcium (58). Thus, GPRC6A may account for some of the effects of strontium on bone cells.
Strontium in Osteoblast-to-Osteoclast Cytokine Signaling
Not only does strontium ranelate act distinctly on bone-forming and bone-resorbing cells, as discussed above, but it also acts by regulating cytokine signals and interactions between these cell types. Specifically, osteoblasts and their precursor cells produce a cytokine, known as the receptor activator of NF-κB ligand (RANKL), that is recognized by a cell surface receptor (i.e., RANK) expressed by osteoclasts and their precursor cells. Activation of RANK through binding of the RANK ligand (RANKL, also known as osteoclast differentiation factor [ODF]) activates intracellular signaling events that result in osteoclast differentiation. Osteoblast cells also release osteoprotegerin (OPG), which binds RANKL and thereby acts as a decoy receptor to prevent the RANK activation; OPG thus functions to inhibit osteoclast differentiation (59). In human primary osteoblasts, strontium ranelate not only increases the expression of OPG, but also decreases the expression of RANKL, doubly undermining osteoclastogenesis (20, 60, 61). In both of these instances, moreover, strontium ranelate exerts its effects upon osteoblastic cells through activation of the CaSR (61). In this way, strontium ranelate acts directly on osteoblasts via the CaSR to promote cell replication, function, and survival, and additionally regulates osteoblast–osteoclast signaling by increasing the OPG/RANKL ratio. Thus, the apparently divergent effects of strontium ranelate—promoting osteoblast function and inhibiting osteoclast function—in fact emanate from the central role of strontium as an activator of the CaSR (Figure 1).
Role of NFAT and Wnt Signaling
An additional mechanism of strontium ranelate action on bone-forming cells has been found to involve nuclear factor of activated T cells (NFAT) transcription factors (62). In basal conditions, NFAT proteins are highly phosphorylated and reside in the cytoplasm. A low, sustained increase in intracellular calcium concentration leads to activation of the cytoplasmic phosphatase calcineurin (Cn) (62), for which the NFAT proteins are substrates. The dephosphorylation of these proteins results in their translocation into the nucleus, where they regulate specific target genes (63, 64). It has become apparent that the Cn/NFAT signaling pathway is an important regulator of bone formation, resorption, and bone mass (65–71). Notably, Cn/NFAT signaling is a positive regulator of osteoblastogenesis, both in vitro and in vivo (72–77).
Intriguingly, strontium ranelate activates Cn/NFAT c1 signaling in osteoblasts (78) although the mechanism for this activation is not completely understood. One possibility is that the activation of the CaSR by strontium (discussed above) may evoke Cn/NFAT c1 signaling via increased intracellular calcium levels (79). In any case, the activation of Cn/NFAT c1 that is observed in response to strontium ranelate further leads to osteoblast cell replication and osteoblast-specific gene expression (78). In particular, recent studies indicate that strontium ranelate activates Wnt signaling in osteoblasts.
Wnt signaling plays an important role in the control of cell replication, differentiation, and survival. Both canonical (β-catenin-dependent) and noncanonical (β-catenin-independent) signaling pathways are involved in the control of bone formation and resorption (80–84). Canonical signals involve Wnt binding to coreceptors LRP5 and Frizzled and culminate in the translocation of β-catenin into the nucleus and activation of target genes (85). Noncanonical signals are transduced through Frizzled and coreceptors such as Ryk, which activate small G proteins including RhoA and ultimately lead to recruitment of NFAT signaling cascades (86). Recent studies indicate that strontium ranelate activates both canonical and noncanonical Wnt signaling in osteoblasts, which in turn positively modulate osteoblast replication and function (78). This recent literature provides evidence for the implication of Wnt signaling in the regulation of osteoblastogenesis by strontium ranelate. Thus, at least two molecular mechanisms of strontium ranelate, one involving Cn/NFAT signaling and the second involving components of Wnt signaling, converge to activate osteoblastogenesis (Figure 3).
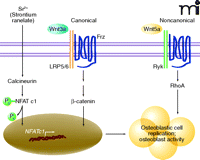
Strontium ranelate activates components of canonical and noncanonical Wnt signaling in osteoblasts. Strontium ranelate activates the calcineurin/NFAT c1 signaling pathway in osteoblasts. This results in increased expression of Wnt proteins (Wnt3a and Wnt5a). Wnt3a interacts with LRP5 (or LRP6) and Frizzled (Frz) coreceptors to activate canonical Wnt/β-catenin signaling. Wnt5a interacts with the Ryk coreceptor to induce noncanonical RhoA-dependent signaling. These two pathways converge to promote osteoblast replication and function.
Preclinical and Clinical Assessments of Strontium Ranelate
The primary mode of action of strontium ranelate in vitro is to oppose bone resorption and formation (87). One important question thus becomes whether this effect, identified in vitro, relates to the therapeutic effect of strontium ranelate in vivo. Among several distinct models of osteopenia, strontium ranelate was shown to exert differential effects upon bone resorption and bone formation and thereby to prevent the induction of bone loss that is associated either with estrogen deficiency, short-term animal immobilization, or high rates of bone turnover (88–91). The positive effects on bone mass and architecture in animal models, however, could not be evoked at low doses (6- to 37-fold lower effective exposure) of strontium ranelate administered to osteopenic rats (92). Efficacy upon bone cells is thus dependent upon appropriate dosage (93).
In keeping with the demonstrated positive role of strontium ranelate on bone mass in osteopenic animal models, strontium ranelate was recently found to increase bone strength and restore microarchitecture in experimental models of implant osseointegration (94). Accordingly, strontium ranelate was also shown to enhance screw fixation in ovariectomized rats (95). Further studies indicate that treatment with strontium ranelate can promote fracture healing, as the result of increased callus and bone formation, in ovariectomized rats (96, 97).
The effectiveness of strontium ranelate as a treatment for osteoporosis is now well documented in a wide range of patients (10, 11, 98, 99). However, some questions remain as to the actual mechanisms involved in the positive effects of strontium ranelate on bone strength. Histomorphometric analysis of bone from patients treated with strontium ranelate has revealed that the compound may act by inducing opposite effects on bone resorption and formation (100); however, this observation remains to be confirmed in studies of control-paired bone biopsies. Another issue is to determine whether the observed positive effect of strontium ranelate in human subjects is due to cellular and molecular mechanisms of action beyond those outlined above, namely, the signal transduction reliant upon CaSR, RANKL/OPG, and NFAT/Wnt pathways. Indeed, preclinical studies suggest that part of the beneficial effect of strontium ranelate on bone strength in vivo may relate to improved bone matrix properties (101). This important aspect of bone quality, as a function of strontium ranelate action, needs to be further investigated in preclinical and clinical contexts. Given the effectiveness of strontium ranelate upon activities as complex as those involved in bone remodeling, it is likely that additional pathways will be implicated in drug efficacy.
Conclusion
The discovery that strontium induces opposite effects on bone resorption and formation in animals continues to be translated into the development of this antiosteoporotic drug. Significant improvement has been made in our understanding of the mechanisms of action of strontium ranelate in bone-resorbing and bone-forming cells. This review has summarized the current knowledge of cellular and molecular mechanisms by which strontium ranelate induces opposing effects on osteoclasts and osteoblasts. Compelling lines of evidence indicate that this distrontium salt acts on osteoclasts and osteoblasts by activating the CaSR, modulating RANKL/OPG, and activating NFAT/Wnt signaling. However, remaining questions concern whether other molecular targets are involved. The identification and relevance of other mechanisms of action to the therapeutic action of strontium ranelate in bone is an important experimental therapeutic problem. Besides bringing clinical benefit to a large and growing population of osteoporotic patients, strontium ranelate may well find therapeutic application in other skeletal disorders.
Acknowledgments
The author thanks past and present members of his group (Monique Hott, Olivia Fromigué, Eric Haÿ, Alain Barbara) who contributed to the experimental studies discussed in this review. This work was supported in part by Inserm, University Paris Diderot, and the Institut de Recherches Servier.
- Copyright © 2010
References

Pierre J. Marie, PhD, DSc, is currently Director of Research at CNRS (Centre National de la Recherche Scientifique). He is Head of the Osteoblast Biology and Pathology Group at Inserm Unit 606 and University Paris Diderot, Paris, France. The focus of his research is on the control of bone formation in metabolic, physical, and genetic bone diseases, and the effects of strontium on bone. E-mail pierre.marie{at}inserm.fr; fax (+33) 1 49 95 84 52.

