Ceramide-1-phosphate: The “missing” link in eicosanoid biosynthesis and inflammation
Abstract
It has been over a decade since ceramide kinase (CERK) and its product, ceramide-1-phosphate (C1P), were first reported. Since itscloning, in 2002, CERK has been the subject of an explosion of publications concerning various signal transduction pathways. The roles of this previously overlooked enzyme, as well as those of its product C1P, continue to expand, and their regulatory functions in the production of eicosanoid inflammatory mediators are proving essential to fundamental signal transduction pathways. In particular, C1P is required for the activation and translocation of cPLA2α, the initial rate-limiting step of eicosanoid synthesis. The potential for inhibitors of CERK to offer a new generation of anti-inflammatory and anti-cancer therapeutics is especially deserving of further study.

Introduction
Ceramide is the central lipid in the metabolism of sphingolipids, and is biosynthesized de novo through condensation of serine and palmitoyl-CoA by the enzyme serine palmitoyl-CoA transferase (1–6). Ceramide is also produced through hydrolysis of sphingomyelin by sphingomyelinases, usually in response to apoptotic agonists (e.g., TNFα) (1–6). These pathways, as well as the mechanisms of regulating ceramide generation, are discussed in detail in several recent reviews by Hannun and colleagues (1–6). For the purpose of this review, the conversion of the core sphingolipid, ceramide, to C1P via the action of the enzyme ceramide kinase (CERK) is of key importance.
CERK was first described as a lipid kinase found in brain synaptic vesicles, in 1989, by Bajjalieh and coworkers (7). This research group demonstrated this lipid kinase activity to be specific for phosphorylating ceramide (i.e., conversion to C1P) but not the closely related glycerol lipid diacylglycerol (7). Just months after this initial finding, Kolesnick and coworkers reported the existence of C1P in human leukemia (HL-60) cells (8), and soon thereafter they reported a CERK activity distinguishable from diacylglycerol kinase (DGK) activity, also in HL-60 cells, in accord with the findings of Bajjalieh and colleagues (9). CERK activity has also been demonstrated in neutrophils (10). To date, CERK-catalyzed phosphorylation of ceramide is the sole established source of C1P in mammalian cells (Figure 1⇓).
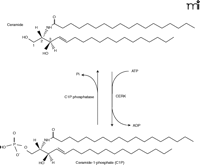
Conversion of ceramide to ceramide-1-phosphate by CERK. Ceramide kinase (CERK) phosphorylates ceramide at the C1 position to produce the signaling molecule ceramide-1-phosphate (C1P). Dephosphorylation of C1P can occur by the action of a specific phosphatase.
All known forms of CERK phosphorylate ceramide specifically at the C-1 position (i.e., without production of ceramide-3-phosphate); CERK also appears to be exclusively membrane-associated, stimulated by calcium, and optimally active at neutral (7, 9–11). CERK is a distinct but close relative of sphingosine kinases; its cloning, which has helped to uncover new physiological functions for C1P, was in fact dependent on its homology to sphingosine kinase type 1 (SphK1) (12). Here, we overview the cellular functions regulated by CERK with a focus on the emerging role and mechanisms of action of C1P in inflammatory responses.
Emerging Roles of CERK and C1P
The first report of a biological effect mediated by C1P came from Gomez-Munoz and coworkers, who demonstrated that short-chain C1P, which is not naturally found in cells, induces DNA synthesis in Rat-1 fibroblasts (13). Subsequently, Gomez-Munoz et al. demonstrated that treatment of T17 fibroblasts with natural (i.e., long-chain) C1P induces a potent increase in DNA synthesis as well as proliferating nuclear antigen (14). These studies also demonstrated that C1P has no effect on mitogen-activated protein (MAP) kinase phosphorylation, c-Myc and c-Fos expression, and phospholipase D activity (14). Thus, the mechanism by which C1P induces DNA synthesis is currently unknown. Other reports indicate that C1P blocks the apoptosis of otic vesicles in response to serum withdrawal (15), and C1P treatment of osteoblastic cells induces the phosphorylation of ERK2 (16).
In the same report describing the existence of neutrophil CERK, Shayman and colleagues demonstrated that CERK itself is activated during phagocytosis after challenging neutrophils with formyl peptide and antibody-coated erythrocytes (FMLP/EIgG) (10). CERK is rapidly activated in this manner, achieving maximum activity within ten minutes. Furthermore, C1P induces liposome fusion to cell membranes, as is demonstrated either through exogenously added C1P or treatment of neutrophils with sphingomyelinase D purified from the venom of the Brown Recluse Spider (Loxosceles reclusa) (10). A more molecular approach, also explored by Shayman et al., verifies the role of CERK and C1P in phagocytosis. In contrast, Rile and colleagues report that C1P levels are not increased in response to cell stimulation in neutrophils (17). Despite this discrepancy, a biological role for CERK in the phagocytosis pathway of neutrophils remains an exciting hypothesis, which is borne out by the activation of cPLA2α by C1P, discussed later in this review.
Intriguingly, CERK is a highly conserved lipid kinase, found not only in animals, but also in plants. In Arabidopsis thaliana, the product of the ACCELERATED CELL DEATH 5 (ACD5) gene is in fact a CERK (18). Plants with an ACD5 mutation initially develop properly but eventually undergo spontaneous cell death (18). Furthermore, mutation of ACD5 results in plants that are more susceptible to infection by Pseudomonas syringae and the acd5 lesion results in accumulation of ceramide, followed by plant shriveling and death (18). Therefore, an important role of plant CERK might be to remove excess ceramide, keeping the organism healthy and resistant to environmental stress. These are intriguing findings that correlate with the proliferation signal mediated by C1P in mammalian cells. Recently, treatment of bone marrow derived–macrophages with C1P has been reported to protect the cells from apoptosis otherwise induced by withdrawal of macrophage-colony stimulating factor (19). Most significantly, these reports collectively indicate that CERK is cytoprotective, in both plant and animal cells, and functions to convert pro-apoptotic ceramide into pro-survival C1P.
The Role of CERK and C1P in the Formation of Eicosanoids
Eicosanoids (e.g., prostaglandins and leukotrienes) are well-established mediators of inflammatory responses, with roles in the pathogenesis of pulmonary infection (i.e., innate immunity), asthma/airway hyper-responsiveness, cancer, atherosclerosis, thrombosis, osteoarthritis, and Alzheimer disease (20–41). The formation of arachidonic acid (AA), via the activation of a phospholipase A2, is the initial, rate-limiting step in eicosanoid biosynthesis (22). In many cases, inflammatory agonists induce activation and translocation of group IVA cytosolic phospholipase A2 (cPLA2α) from the cytosol to perinuclear membranes; however, the mechanism and mediators of cPLA2α translocation have not been completely elucidated (42). Consequently, cyclooxygenase 2 (COX-2) and other enzymes of eicosanoid metabolism are induced (42). COX-2 levels in most cases increase prior to cPLA2α activation, effectively “priming” the system for optimal response. COX-2 then utilizes the AA released by cPLA2α to initiate the prostaglandin synthetic pathways; in the leukotriene pathway, the initial enzyme, lipoxygenase (LO), similarly utilizes AA as a substrate (42).
Implications for ceramide as a signaling molecule for inflammatory responses come from a variety of contexts. A cell-permeable ceramide analog affects prostaglandin production, for example, whereas ceramide itself—or sphingomyelinase, in a dose-dependent manner—enhances IL-1–mediated prostaglandin E2 (PGE2) production (43). Subsequently, it was demonstrated that ceramide acts synergistically with the Ca2+ ionophore A23187 to cause the translocation of cPLA2α, although ceramide alone has no effect (44–46). It was thus suggested that ceramide regulated eicosanoid synthesis via cPLA2α; however, recent studies have shown that C1P, rather than ceramide, functions as a proximal mediator of AA release (47).
Several lines of evidence support the concept that C1P is the active agent in signaling through cPLA2α activity. First, unlike ceramide, exogenous C1P alone can stimulate AA release and PGE2 formation (47). Second, treatment of cells with spider venom sphingomyelinase D to produce C1P from membrane sphingomyelin can elicit the AA response, whereas sphingomyelinase C, which produces ceramide, has no effect (47). Third, inflammatory agonists, such as IL-1β, increase endogenous C1P levels within the proper time frame to account for AA release (47). Importantly, the downregulation of CERK by use of RNA interference technology (RNAi) dramatically interferes with the ability of ATP, A23187, and IL-1β to elicit AA release and PGE2 synthesis (47). Finally, PLA2α activity is required for C1P to promote AA release (48). C1P, and not ceramide, is thus demonstrated to act as the upstream signal for eicosanoid synthesis (47) and as a regulator of inflammatory responses (Figure 2⇓). Thus, the earlier observations of the synergistic effects of ceramide and inflammatory agonists were likely due to conversion of ceramide to C1P, suggesting that CERK is activated in response to these inflammatory agonists (e.g., A23187 and IL-1β).
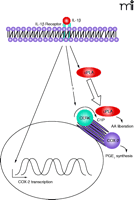
Role of CERK in eicosanoid synthesis. In response to inflammatory agonists, such as interleukin-1β (IL-1β), the cell is “primed” for eicosanoid synthesis through induction of cyclooxygenase-2 (COX-2). CERK then plays a pivotal role by producing C1P, a signaling molecule required for effective translocation (as indicated by the block arrow) of cPLA2α to the perinuclear membranes and consequential generation of AA. Conversion of AA to prostaglandin E2 (PGE2) represents the effective outcome of the inflammatory agonist. The question mark indicates that further signaling steps my prove to recruit CERK activity directly. Straight line arrows represent positive modulation of signal transduction; curved arrows indicate enzymatic reactions.
C1P was recently reported to regulate the degranulation of mast cells, important players in inflammatory responses (49). Mitsutake and coworkers demonstrated that overexpression of CERK enhances degranulation of RBL-2H3 cells induced by A23187 (49). Because the interconversion of C1P with the other bioactive sphingolipid metabolites was not considered in this study, however, it remains possible that the active agent contributing to degranulation could be sphingosine-1-phosphate (S1P). This possibility is of key importance, because the generation and secretion of S1P, which in turn activates the S1P2 receptor on mast cells, also induces degranulation (Figure 3⇓) (50, 51). Establishing whether S1P and C1P have non-overlapping roles in this case therefore warrants more comprehensive studies, as is underscored by recent unpublished findings demonstrating that C1P induces PGD2 synthesis in RBL-2H3 and LAD2 cells without effect on the degranulation mechanism of these cell types (Figure 2⇑; C Chalfant, unpublished observations).
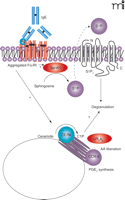
SphK1 and CERK in mast cell functions. Crosslinking of FcεRI, the high-affinity IgE receptor on mast cells, results in translocation (to the cell membrane) and activation of sphingokinase-1 (SphK1). The resulting product, S1P, in turn activates S1P2 receptors, leading to mast cell degranulation. CERK may also regulate degranulation of mast cells.
The Complex Regulation of CERK
These recent studies go a long way toward clarifying the complex body of actions attributed to C1P in inflammatory processes, phagocytosis, and cell survival mechanisms; however, the mechanisms by which CERK is activated by cellular agonists (e.g., IL-1β) requires further elucidation. Thus, several important questions remain, especially concerning the regulation of CERK and the intracellular targets of C1P signals.
As for the first matter, almost nothing is known as to how CERK is activated in response to cellular agonists, but the recent cloning of CERK from Jurkat acute T-cell leukemic cells (52) will facilitate structure–function characterization of the enzyme. The ectopic expression of CERK in HEK-293 cells fortunately recapitulates the biochemical characteristics of the enzyme first described in brain extracts (7, 11, 52), which will also allow for further characterization. Human CERK, mainly expressed in heart, kidney, brain, and hematopoietic cells, occurs as a 537-residue protein closely related in structure and amino acid homology to the sphingokinases, containing the five conserved domains (C1–C5) previously identified in SphK1 and SphK2 (52). The first three of these domains, C1–C3 (i.e., amino acids 126–278), comprise a putative diacylglycerol kinase domain (DGK domain) (52) (Figure 4⇓). Sugiura and coworkers have suggested that the specificity of CERK for ceramide, with no activity toward sphingosine, may arise from subtle changes in the amino acid sequence of the C1, C2, and C3 regions that are observed in comparison to the corresponding sequences of SphK1 and SphK2 (52). CERK also appears to contain several regulatory regions (52), such as a calcium/calmodulin binding motif of the 1-8-14 type B spanning residues 422–435. In this regard, Igarashi and coworkers recently reported that calmodulin is involved in the calcium-dependant activation of CERK, acting as a “calcium sensor” for the enzyme (53). Their data suggest that increased addition of calmodulin stimulates CERK activity, even in the presence of low of calcium concentrations. Furthermore, mutation of the calmodulin binding site significantly affects the activation of CERK, in vitro and in cells, by A23187. This observation concurs with another recent publication, from Van Veldhoven and colleagues, which reports that CERK expressed in bacteria (i.e., not associated with calmodulin) undergoes activation by calcium in the micromolar range and not the nanomolar range as previously observed for the partially purified enzyme (54). Thus, calmodulin likely regulates the calcium sensitivity of CERK, and several reports, using standard radiolabeling techniques, have shown that the calcium ionophore A23187 stimulates C1P formation in various cell types (47, 55–57). Unpublished findings from our laboratory also confirm this observation using mass spectrometry technology. With calcium/calmodulin thus implicated in the regulation of the enzyme, CERK/C1P may well have roles in signal transduction mechanisms (e.g., eicosanoid synthesis) that utilize calcium influx.
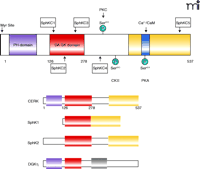
The structure of CERK. The structural domains of CERK include the PH domain (purple), the diacylglycerol kinase homology domain containing subdomains C1 through C3 (red), and the C-terminal domain (yellow) containing the C5 subdomain and the calmodulin interaction site (blue). Conservation of some of these domains is reflected among other kinases, including sphingosine kinase 1 (SphK1), sphingosine kinase 2 (SphK2), and diacylglycerol kinase η (DGKη). Modification sites within the CERK structure include a casein kinase II phosphorylation site (CKII) at Ser340, a cAMP-dependant phosphorylation (PKA) site at Ser424, and a protein kinase C (PKC) phosphorylation site at Ser300.
Another plausible mechanism of regulating CERK activity is through lipid interactions. For example, almost all of the CERK (i.e., ninety-nine percent of activity and protein) found in cells is associated with the membrane fraction. Furthermore, the molecular cloning of CERK identifies a pleckstrin homology (PH) domain, at the N terminus of CERK (12), known to bind phosphoinositol-4,5-bisphosphate (52), and this PH domain has been shown to be essential for the localization of CERK to specific membranes in cells (Figure 4⇑) (58). Carre and coworkers report that overexpression of GFP-tagged CERK in COS1 cells results in localization to perinuclear/Golgi membranes (58), and this same pattern of distribution occurs in A549 adenocarcinoma cells for both recombinant and endogenous CERK (C Chalfant, unpublished work). Other studies, by Igarashi and coworkers, show that CERK localizes mainly to the cytosol in RBL-2H3 cells (49). Currently, the cause of the differences observed in the subcellular localization of CERK is unknown, but these contrasting reports may reflect variations in cell types or fixation methods. Common to all studies was the necessity of the PH domain for proper localization in cells; alteration of specific residues in this domain can lead to aberrant intracellular localization of the enzyme (58). In addition to the PH domain, other undiscovered lipid interaction domains are also required to localize the enzyme to the proper cellular organelles.
The PH domain of CERK is also required for catalytic activity in that its removal results in a catalytically inactive protein (58). Furthermore, Igarashi and coworkers have demonstrated that mutation of Leu10 in the PH domain abolishes CERK activity, suggesting that this amino acid is essential to catalysis (59). Because all catalytic activity is lost by either specific mutation or deletion of the entire PH domain, structural effects may explain these observations.
The specific lipids that interact with CERK are unknown, but cardiolipin has been recognized for over a decade cofactor for CERK (7). Little structural information is available concerning specific interaction(s) between CERK and cardiolipin, and so similarly, cardiolipin’s role in supporting CERK activity is poorly understood. The PH domain may be involved in the interaction, but the enzyme does not localize to the mitochondrial membrane, where cardiolipin is enriched. These data suggest that a closely related lipid cofactor (e.g., Bis-lysophosphatidic acid) enriched in the Golgi apparatus may possibly be a lipid cofactor for CERK. Future studies will hopefully answer this conundrum of activation versus localization.
Another possible mechanism of increasing C1P in membranes in response to cellular agonists is via transcriptional regulation of CERK. A recent report from Euskirchen et al. suggests that CERK is upregulated in response to phosphorylation of the cyclic AMP response element-binding protein (CREB) and identifies a CREB binding site on the promoter region (60). Thus, increased expression of CERK may be a key mechanism of increasing C1P levels in the cell if substrate (i.e., ceramide) is available. Additionally, CERK appears to be a phosphoprotein (61), and in fact CERK contains two phosphorylation sites conserved across several species, a casein kinase II phosphorylation site [(S/T) XX (D/E)] at Ser340 and a cAMP-dependent phosphorylation site at Ser424 (52) (Figure 4⇑). There are also six putative mammalian protein kinase C (PKC) phosphorylation sites [Ser72, Thr118, Thr127, Ser230, Ser300 (also conserved in SphK), and Ser340] (52). Phosphorylation has been shown to regulate the closely related enzyme SphK. More research into each of these regulatory mechanisms will be required to understand the activation of CERK in response to specific agonists.
CPLA2α: A Direct Target of C1P
Unlike S1P, C1P is not thought to act through a cell surface receptor but rather likely functions at the intracellular level. To elucidate the direct intracellular target(s) for C1P in response to inflammatory agonists, cPLA2α was an obvious object of investigation, because treatment of various cell-types with C1P induces the release of free AA. Furthermore, cPLA2α has a calcium-dependent lipid binding (CaLB) domain similar to the C2 domain of protein kinase C (PKC) (21). Thus, C1P could be envisioned to interact directly with this domain in a fashion analogous to the interaction of phosphatidylserine with the C2 domain of PKC. Alternatively, C1P could indirectly activate cPLA2α by mobilizing calcium (62). We have recently discovered that C1P is indeed a direct activator of cPLA2α through interaction with its C2/CaLB domain (63). This finding contradicts current dogma that zwitterionic lipids, such as phosphatidylcholine, rather than anionic lipids such as C1P, bind to the C2/CaLB domain (64). Nevertheless, there is significant interaction between C1P and cPLA2α, particularly through the C2/CaLB domain at 300 nM calcium (63), which is in intriguing agreement with early suggestions that association of cPLA2 with membranes requires 300 nM calcium (21).
Importantly, C1P specifically activates cPLA2α both by an allosteric mechanism and by lowering the dissociation constant of the enzyme (and C2 domain) from phosphatidylcholine-rich vesicles (63). Given the latter effect, C1P could be involved in the recruitment of cPLA2α to the Golgi apparatus and perinuclear membranes. Because CERK localizes to the Golgi apparatus and endoplasmic reticulum in HUVECs, COS-1, and A549 cells (65), C1P can be generated in the appropriate cellular compartment for recruitment of cPLA2α in response to inflammatory agonists. We have also found that cPLA2α and CERK colocalize when cells are challenged with inflammatory agonists (C Chalfant, unpublished observation). The culmination of these data demonstrates that CERK and cPLA2α, which share the same tissue distribution, are intrinsically linked. Intriguingly, recent work indicates that C1P interacts with a novel site within the C2 domain of cPLA2α, namely, near calcium binding region II (CBR II), and does not compete with the sites that bind phosphatidylcholine (i.e., CBR I and III) and PIP2 (i.e., the catalytic domain) (63) (Figure 5⇓).
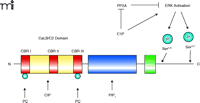
A hypothetical mechanism of cPLA2activation in response to the generation of C1P. The structure of cPLA2α includes a CaLB/C2 domain (red and yellow), containing calcium binding regions (CBRs I, II and III; red), the PIP2 domain (i.e., the catalytic domain; blue), and the hinge region (green). C1P has multiple roles in activating cPLA2α. Within the structure of cPLA2α, Ser505 and Ser727 can be phosphorylated (blue spheres), by ERK, to activate cPLA2α; C1P is shown as a direct activator of ERK as well as an indirect activator of ERK by inhibiting protein phosphatase 2A (PP2A). C1P also interacts directly with cPLA2 near CBRII. Additionally, C1P increases calcium (Ca2+) concentrations, in a compartmentalized manner, in order to induce the association of cPLA2α with perinuclear membranes. Sites of interaction with C1P and phosphatidylcholine (PC) are also indicated. See text for details.
The question remains as to whether the generation of C1P is sufficient to elicit the translocation/activation of cPLA2α or whether calcium mobilization in conjunction with the generation of C1P is required. Clearly, previous findings demonstrate that C1P is required, because downregulation of CERK, resulting in diminished C1P levels, abolishes calcium ionophore–induced AA release and inhibits ATP-induced AA release (66). C1P has been reported to induce calcium mobilization in thyroid FRTL-5 cells, but another recent report demonstrates that C1P does not mobilize calcium in neutrophils (17, 67). Findings from our laboratory are in agreement with the latter report in that C1P did not induce calcium mobilization in A549 cells using a FURA-2 based assay (47, 48). These results nevertheless allow for the possibility that C1P may regulate calcium homeostasis in A549 cells by affecting highly compartmentalized calcium microdomains, where calcium concentrations can be dramatically altered. Because the hydrolysis of sphingomyelin, with subsequent increase in ceramide (i.e., the metabolic precursor of C1P), would affect membrane fluidity, a compartmentalized increase in calcium is certainly possible. Furthermore, a calcium-dependent mechanism of cPLA2α activation by C1P is clearly suggested. Thus, a coactivation mechanism, where C1P raises calcium concentrations in a highly compartmentalized manner, is readily hypothesized. The collaborative actions of C1P and calcium in the activation of cPLA2α are indicated in Figure 5⇑.
The demonstration that cPLA2α and C1P are intrinsically linked in function would explain the observation that cPLA2α and C1P/CERK are reported to regulate similar cellular processes. For example, as we have alluded (see above), C1P plays a role in phagocytosis by neutrophils (10). Interestingly, both Leslie and Gelb and their respective laboratories recently demonstrated a role for cPLA2αin macrophage and neutrophil phagocytosis (37, 56, 57, 68). Thus, as with eicosanoid biosynthesis, cPLA2α and CERK may be linked in phagocytic processes.
Other possible direct targets of C1P are protein phosphatase-1 and -2A (PP1 and PP2A). These enzymes are otherwise known as “ceramide-activated protein phosphatases” (CAPPs) and have been linked to ceramide-induced apoptosis (69). Activation of PP1 by ceramide leads to dephosphorylation of “SR proteins,” so named for their characteristic serine/arginine domains, which modulate mRNA splicing both to reduce levels of the anti-apoptotic protein Bcl-x(L) and to increase levels of the pro-apoptotic Bcl-x(S). Interestingly, C1P is a potent inhibitor (IC50 = 50 nM) of both PP1 and PP2A in vitro (70), which is consistent with the mitogenic/cell survival effects of C1P observed by Brindley and colleagues; inhibition of these protein phosphatases appears to activate the ERK1/2 pathway and increase DNA synthesis (71, 72). The potential of C1P to function as a signaling molecule by inhibiting PP1 activity may also have direct relevance to the role of alternative splicing in cancer. It has been demonstrated that the generation of ceramide induces a pro-apoptotic change in the alternative splicing of caspase 9 and Bcl-x pre-mRNA (69, 73). Importantly, this effect is mediated by ceramide-dependent activation of PP1 (69, 73). Because C1P is a potent inhibitor of PP1, one can hypothesize that C1P generated by CERK may be an antagonist to ceramide action and thus mediate cytoprotective signaling. Accordingly, Gomez-Munoz and coworkers have demonstrated that C1P induces a cell survival/anti-apoptotic effect on macrophages by increasing the expression of Bcl-x(L) (19). Unfortunately, they did not examine the expression of Bcl-x(S). If C1P indeed promotes Bcl-x(L) activity at the expense of Bcl-x(S), then CERK may be a critical switch that opposes apoptotic agonists mediated through ceramide. By analogy to SphK1, which converts pro-apoptotic sphingosine to anti-apoptotic S1P, CERK may thus play a homeostatic role in regulating alternative splicing of Bcl-x pre-mRNA.
Inhibition of PP1 and PP2A by C1P may also have implications in coordinating Ca2+-dependent activation of cPLA2α with another reported mechanism of cPLA2α activation, that is, phosphorylation of Ser505 and Ser727 (74, 75). Phosphorylation of both serine residues is required for both calcium ionophore- and cytokine-induced AA release, and several reports suggest that the MAP kinase pathway is responsible for modulating the phosphorylation state of cPLA2α (74, 75), which correlates well with the report demonstrating that C1P treatment activates the MAP kinase pathway (16). Furthermore, okadaic acid, a small-molecule inhibitor of serine-threonine protein phosphatases, has also been shown to induce AA release via cPLA2αin a calcium-independent manner, possibly through effects on the phosphorylation state of the enzyme (76). Therefore, okadaic acid may be an artificial means of mimicking the intracellular role of endogenously generated C1P in the inhibition of PP2A. The generation of C1P may act indirectly to enhance the phosphorylation of Ser505 and Ser727 of cPLA2 by inhibiting protein phosphatases and thereby activating the MAP kinase pathway.
CERK: A Therapeutic Target?
Given that CERK plays a major role in the regulation of the cPLA2αand AA release, a pathophysiological link can now be inferred between the production of C1P and inflammatory responses mediated by AA and eicosanoids. The demonstration that CERK/C1P generation is upstream of cPLA2α activation suggests that therapeutics that could inhibit CERK could comprise a new generation of therapeutics for inflammatory disorders such as asthma, atherosclerosis, and Alzheimer disease. Therapeutics based on CERK would also have the benefit of blocking AA liberation as well as the unwanted formation of leukotrienes and COX-1 derived prostanoids (e.g., thromboxanes), possibly lowering the problems of side-effects associated with COX inhibitors.
Because of the strong association between chronic inflammation and cancer (77, 78), and in light of the prosurvival function of C1P, CERK may be a pharmacological target for development of novel anti-cancer therapeutics. In this regard, non-steroidal anti-inflammatory drugs are also being used to treat cancer (79–82). Inhibitors of COX-2 have been shown to reduce the growth and metastasis of some cancers (79–82). Specifically, in mouse models of familial adenomatous polyposis, it has been shown that crosses with cPLA2α- or COX-2–deficient knockout mice reduce the size and metastatic capability of tumors (79, 83). cPLA2α knockout mice also demonstrate a large reduction (greater than forty-nine percent) in the size and number of lung tumors produced by urethane injections. Thus, the link between eicosanoids and these two cancers has been validated in vivo, suggesting that CERK inhibitors may have the same effects as removing cPLA2α.
Unfortunately, effective and specific inhibitors of CERK remain elusive. Recently, our laboratory published an in-depth study on the substrate specificity for CERK (84). The goal of this study was to identify moieties in the ceramide molecule required for its recognition and utilization as a CERK substrate. In brief, modification to any of the ceramide moieties greatly reduces CERK specificity for the resulting derivative (84). These findings have obvious implications for the rationale design of competitive/slow-dissociating inhibitors for CERK. Our data suggest that substrate recognition by CERK requires preservation of an acyl chain that includes at least twelve carbon atoms, a requirement that would significantly reduce solubility of the compound and cellular uptake, and thus, efficacy (84). Still, the development of competitive inhibitors of CERK is only one route to an effective and specific inhibitor of the enzyme. CERK activity also requires the association of membranes with the enzyme’s N-terminal PH domain, so that noncompetitive inhibitors might also be rationally designed. In any case, the development of high-affinity inhibitors of this enzyme is warranted in light of the important biological processes regulated by CERK.
In conclusion, CERK and its product, C1P, have been shown to regulate important cellular processes in plants as well as mammals, demonstrating the broad and important role for this enzyme in cell signaling cascades conserved across evolution. We hope that recent exciting and provocative results discussed here will stimulate the scientific community to undertake the daunting tasks of identifying intracellular targets for C1P so that clinically useful CERK inhibitors might be assessed within the contexts of inflammation and cancer.
Acknowledgments
We thank the members of the Chalfant laboratory whose diligent efforts contributed to much of the work cited here. This work was supported by NIH grant R01 HL072925 and a VA MERIT I Award to CEC.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Nadia F. Lamour, PhD, is a postdoctoral fellow in the Department of Biochemistry at Virginia Commonwealth University School of Medicine. Her interests pertain to lipid regulators of inflammation.

Charles Chalfant, PhD, is Assistant Professor of Biochemistry at Virginia Commonwealth University School of Medicine. His interests include the extracellular cues that support cellular programmed death, especially as these relate to the (patho)physiological function of the apoptosis regulator Bcl-x(L) and caspase 9.

