CALCIUM: A Role for Neuroprotection and Sustained Adaptation
Abstract
Sustained increases in intracellular calcium following prolonged seizures or other neurological insults have been thought to be responsible for neuronal cell death for well over two decades. For example, a seizure or a stroke can lead to excessive release of glutamate, an endogenous excitotoxin. Overactivation of receptors that interact with glutamate will raise calcium levels to stimulate a variety of signaling pathways that can impair neuronal respiration and eventually kill neurons. On the contrary, recent evidence shows that under numerous conditions calcium can prevent neurons from dying. Experimental epilepsy and ischemia models show that protection of neurons appears to depend upon the age of the animal, the amount and route of calcium elevation, timing of initial insults, and brain regions involved. This review will discuss novel findings on the protective signaling role of calcium under a wide range of pathological conditions.

Introduction
Sustained calcium (Ca2+) influx through glutamate receptor channels is thought to represent a final common pathway of neuronal cell death that is associated with a number of neurodegenerative diseases such as epilepsy, hypoxia-ischemia, hypoglycemia, Alzheimer Disease, and schizophrenia (1–4). l-Glutamate, the major excitatory neurotransmitter of the central nervous system (CNS), is released in excess in all of these diseases and consequently stimulates Ca2+ release from neuronal stores by exerting its actions on receptor membranes from three structurally related fast acting ionotropic gene families: NMDA (N-methyl-d-aspartic acid), AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid), and KA (kainic acid). l-Glutamate also interacts with metabotropic (mGluR) receptors that mediate slow synaptic responses, owing to their coupling to intracellular G proteins (5). For example, the mGluR1 and mGluR5 subunit subtypes that exist in a number of alternatively spliced forms (mGluR1 α, β, χ, and δ, and mGluR5 α and β), are coupled to inositol trisphosphate (IP3)/ Ca2+ signal transduction and contribute to intra-signaling cascades that may alter downstream pathways such as protein kinase activation and contribute to delayed cell death processes (5). High signal efficacy can be achieved because excess levels of glutamate in the central nervous system can result in elevated intracellular Ca2+ concentrations ([Ca2+]i) that exist for several hours even after glutamate has been removed from the synaptic cleft (6, 7). This increase, in turn, causes a rise in [Ca2+]i in sensitive organelles, such as the mitochondria (8–9) and endoplasmic reticulum (10), whereby membrane-targeted NMDA receptors contribute to the spatio-temporal dynamics of intracellular Ca2+ elevations (11). The molecular events linking glutamate receptor activation and elevated cytosolic free-Ca2+ to eventual cell death include downstream activation of proteases, endonucleases, and the generation of nitric oxide and free radicals, which ultimately destroy cells by lipid peroxidation (3, 12–14), apoptosis (2), or both. Excess release of zinc at mossy fiber synapses—associated with the sustained release of l-glutamate—also contributes to excitotoxicity, preferentially via voltage-gated Ca2+ channels (15). Notably, however, large increases in [Ca2+]i can also suppress cell-death genes to prevent programmed cell death and promote neuronal survival (16, 17). This survival-promoting effect is reportedly mediated by sustained elevations of cytoplasmic free Ca2+ concentrations that are caused by influx of Ca2+ through voltage-gated channels activated by K+-induced chronic depolarization (18).
NMDA receptors are Ca2+-preferred glutamate-gated ion channels that are expressed throughout most central neurons. Their functional properties are determined by the expression of specific splice variants of each subunit and the assortment of these subunits into working channels (19). NMDA-type glutamate receptors were initially held responsible for neuronal injury, owing to their high Ca2+ permeability conductance properties (termed the “excitotoxic hypothesis” or “Ca2+ overload hypothesis”) (20). AMPA-type glutamate receptors, however, have also been implicated in excitotoxicity because these receptor assemblies, which lack the GluR2B subunit, are highly permeable to Ca2+ (21–23) and possibly contribute to the delayed neuronal cell death processes by Ca2+ overload (termed “the GluR2B hypothesis”) (24–27). On the other hand, many additional studies also show that changes in glutamate receptor expression after neurological insults may not be so selective (28–31). It is important to note that the specific AMPA and NMDA receptor patterns expressed after an insult depend upon the age of the animal and history of early-life seizures (32). Therefore, under pathological conditions, such as KA-induced seizures or hypoxia-ischemia, many principal cells may increase their Ca2+ influx regardless of the existing stoichiometry of AMPA or NMDA receptor assemblies. The present article will review how gene mutations, sustained seizures, or other traumas have developmentally regulated effects on glutamate receptors and Ca2+ permeability that may lead to long-term adaptation.
NMDA Receptors, Ca2+ Permeability, and Delayed Cell Death
NMDA receptor subunits (e.g., NR1, NR2A-D) are expressed in distinct spatial and temporal patterns throughout development, suggesting that the receptor subunits may enable unique functions. These receptors are critical for neural plasticity, normal development of the nervous system, and survival of the organism. Mice deficient in the NR1 gene lose classical NMDA electrophysiological responses and these mutants die as neonates (33). Even a single point mutation within the NMDA receptor channel can cause a number of neurological phenotypes, signifying how critical the NMDA receptor complex is for modifying neuronal development and plasticity (34). Cell survival mechanisms were also revealed by pharmacological blockade of NMDA receptors with channel antagonists such as phencyclidine (PCP), which causes corticostriatal neuronal death (35). Moreover, genetic deletion of NR1 or NR2B subunits in mice prevents the appearance of whisker representations (sensory mapping) in the somatosensory corticothalamic system (33, 36, 37) and kills neurons in the developing somatosensory thalamus (38). Thus, neuronal growth and survival appear to depend on the quantity of glutamate that modulates Ca2+ influx via NMDA receptors, resulting in the expression of “early” cell death genes or the activation of signal cascades critical for cell survival and synaptogenesis (39–41).
The general assumption that altered Ca2+ permeability kills neurons via glutamate receptors such as NMDA- or AMPA-types is oversimplified. This is because susceptibility to seizures by certain convulsion-eliciting compounds, hypoxia, or other trauma-induced injury, depends upon a number of factors. For example, excitotoxic and seizure effects vary with dosage (e.g., of NMDA or KA), animal species or strain undergoing the trauma, the severity of the insult, the timing of insults, and brain regions involved (42–52). High doses of NMDA (10 μM) microdialysed into the sensorimotor cortex depress local electroencephalograph (EEG) activity but evoke epileptogenic seizures in the hippocampus under the same conditions (48). In adult rats, high doses of NMDA (25 nmol) induces clonic seizures when delivered to the thalamus mass intermedia (46) or when delivered to the lateral ventricles of adult mice at low doses (2 nmol) (47). Both high and low doses produce damage in adult hippocampal neurons with different time courses; however, our recent data show postnatal day 13 (P13) pups are susceptible to seizures but neurons are not damaged at low doses of NMDA (2.5 nmol) (53). The anatomical location of NMDA administration is also critical as high intrahippocampal doses of NMDA can result in partial delayed damage of the CA1 but the amount of this injury is much less than that reported in neonatal rat pups given high doses of NMDA (54); or pontosubicularly administered NMDA (25 nmol) (55).
Contrary to our expectations, reduced expression (i.e., knockdown) of the NR1 subunit [accomplished with antisense oligonucleotides (AS-ODNs)] reduced NMDAR-mediated Ca2+ permeability but elicited a preferential neurotoxicity to the CA1 subregion after rat pups were given subtoxic doses of NMDA or after KA was injected peripherally to induce seizures. These findings suggest that high constituent amounts of NR1 subunits are required for survival of CA1 neurons during the second postnatal week of rodent development. Immature CA1 neurons may become particularly vulnerable after knockdown of the NR1 subunit. CA1 sensitivity might arise from simultaneous loss of NMDA receptors expressed on GABAergic interneurons that lead to disinhibition of GABAergic inhibitory neurons, resulting in a low-grade chronic excitotoxicity in accordance with the “hypofunction hypothesis of schizophrenia” (56).
AMPA Receptors, Ca2+ Permeability, and Delayed Cell Death
AMPA-type glutamate receptors exist as Ca2+-permeable and Ca2+-impermeable channels (21–23, 57). Ca2+-permeable AMPA receptors were thought to participate in seizure- and ischemia-induced hippocampal damage because GluR2B mRNA and protein expression were selectively decreased in vulnerable CA1 or CA3 neurons before cell death (24, 25, 27, 58) but were sustained in immature neurons resistant to seizure-induced brain damage (59). However, less selective changes in glutamate receptor expression after neurological insults have also been reported (28–31). It is possible that alterations in gene expression may simply reflect adaptive changes in the amount of depolarization achieved by AMPA receptor activation (60), which is likely because of the wide diversity of endogenous AMPA receptors whose subunits exist in differing stoichiometries at different brain sites (22).
Kainic acid (KA), an analog of glutamate and a potent neurotoxin (61), is widely used to study neurodegeneration in adult animals because systemic or intrathecal injection induces persistent synchronous electroencephographic (EEG) alterations and marked neurodegeneration of the hippocampus and other limbic structures that closely resemble human temporal lobe epilepsy (62). Interestingly, the immature brain is relatively resistant to hippocampal lesions after experimentally induced status epilepticus until a critical stage in development around third postnatal week; however, the immature brain is highly sensitive to hippocampal damage after neonatal ischemia (60, 62, 63). Both traumas (i.e., status epilepticus and ischemia) result in large increases in [Ca2+]i in the immature hippocampus, but neuronal cell death is only observed under severe anoxic conditions. Induced expression of proapoptotic Bcl-2 family members, death receptors, and caspases were associated with cell death after neonatal ischemic injury (64) but not after neonatal seizures to explain the above potential differences (65). Not only does KA have different effects on hippocampal vulnerability in neonates vs adults, as observed after NMDA application, but recent evidence from our laboratory showed that patterns of neuronal damage (Figure 1A⇓) and expression of AMPA receptors (Figure 1B⇓) after KA-induced status epilepticus also depend upon the maturational stage of the animal and history of early-life seizures (32, 45). In adult rats, extensive neuronal injury of CA3 neurons—as marked by silver stain but without obvious cell loss—occurred at 16–24 hours after status epilepticus; obvious CA3 lesions were present and near complete at forty-eight hours (59, 66) (Figure 1A⇓). In contrast, at juvenile ages (P20 and P30), neuronal injury of the hippocampus did not appear until after seventy-two hours and was restricted to the cell body of CA1 pyramidal neurons. At these immature ages, many CA1 neurons were injured (histological observation) but few or no cells were actually lost after single or multiple injections of KA (45) (Figure 1A⇓). Thus, there are age-dependent patterns of hippocampal damage after a single injection of KA (1×KA). CA1 neurons may be more susceptible to excitotoxic injury in the juvenile period owing to excitatory properties of GABA receptors (67) and insufficient development of GABAergic inhibitory inputs to their dendrites (52).
Because a maturational switch of neuronal vulnerability from CA1 to CA3 occurs with age (45, 69), age-dependent differences in gene and protein expression were also anticipated to occur in the juvenile period after single vs multiple episodes of status epilepticus. Contrary to studies in adult rats where the GluR1A/GluR2B ratio was increased (25, 27), recent findings have demonstrated that after a single prolonged seizure, juvenile animals undergo a significant reduction in the GluR1A/GluR2B ratio that is specific to the injured CA1 neuronal population (Figure 1B⇓), similar to the increased GluR2B expression evident in CA1 (twenty-four hours post-ischemic tolerance) (70). Another inconsistency with AMPA receptor–mediated Ca2+ permeability as being responsible for cell death is that in a number of models of delayed neuropathology, marked downregulation of the GluR2B subunit also persists in resistant cell types (Box 1). Finally, a history of early-life seizures rescued the GluR1A/GluR2B ratio in the CA1 to control values, suggesting that the two earlier seizure insults reduced the impact of the third insult even after a long delay (from P13 to P30) (45). NMDA receptor subunit composition was relatively resistant to change in the hippocampal subregions after single or multiple seizures in the juvenile period, similar to results obtained in receptor mRNA studies performed in adults (25, 32). In contrast, long-term decreases in both GluR1A and GluR2B subunits were observed in adulthood without cell loss after three (71) or after only one episode of status epilepticus induced in the neonatal period (72). Therefore, the previously proposed synergism of decreased GluR2B expression and cell death is inconsistent with the increase of GluR2B expression in the juvenile period after status epilepticus, with low or no expression in resistant cell types, and also with sublethal ischemia models. It appears that the timing of insults is critical to clinical outcomes such that a single insult may be neurotoxic after a critical age, whereas multiple insults may be neuroprotective if seizures begin early in life.
High Ca2+Permeability in Resistant Cell Types
Increases in Ca2+ permeability are thought to occur after a neuropathological insult in neurons destined to die (134). However, increases in Ca2+ permeability also occur in resistant cell types normally lacking the GluR2B subunit (135). AMPA receptor-mediated toxicity has also been implicated in selective neuronal cell death associated with amyotrophic lateral sclerosis (136, 137). However, quantitative measurements of relative Ca2+ permeability of AMPA receptors in healthy motor neurons show that all motor neurons have intermediate whole-cell Ca2+ permeability and that the relative abundance of GluR2B mRNA varies more in dorsal horn sensory neurons, which are resistant to damage (137). Similarly, AMPA receptor subunit expression studies carried out after an experimental spinal contusion injury, show persistent and selective decrease expression of the GluR2B subunit in surviving spinal cord tissue resistant to injury (138). After induction of glaucoma in primates, both NMDA and GluR2B proteins were reduced in retinal ganglion cells vulnerable to death and both were unchanged in less vulnerable populations, such as bipolar horizontal and amacrine cells (139). This suggests that Ca2+ permeability, per se, is not the only factor responsible for cell death. In addition, after unilateral retinal ischemia and forty-eight hours of reperfusion, AMPA receptor and NR1 subunit immunoreactivities are either increased or sustained in the retina and not decreased in the vulnerable ganglion cell layer (140). Thus, a number of models of delayed neurodegeneration imply that persistent downregulations of glutamate subunit types that permit increases in Ca2+ permeability also occur in surviving cells of the retina, hippocampus, and spinal cord. In keeping with this, many interneurons that lack the GluR2B are vulnerable after a seizure or ischemic insult, further reducing a correlative relationship between changes in AMPA receptor expression with cell death. In cultured Purkinje cells, substantial increases in [Ca2+]i occur even in neurons that contain high levels of GluR2B subunits when kainate (KA) exposure is prolonged (141). It is proposed that adaptation may arise by the changes in glutamate receptor assemblies in the surviving populations and those changes in the surviving populations may be responsible in altering subsequent responses to l-glutamate and synaptic plasticity.
Induced Expression of Ca2+-Permeable AMPA Receptors
Advancement of transgene methodologies (73, 74), phosphodiesterized AS-ODN knockdown approaches of the GluR2B subunit (75–77), and development of ischemia tolerance studies (70) support a role for Ca2+ in “excitotoxic-induced adaptation” (Box 2). Genetic targeting of a critically located arginine residue (Q/R editing site, position 586 within the channel pore) which is subjected to posttranscriptional editing and consequently responsible for gating of divalent ions of the mature GluR2B subunit was modified (78). Replacement of the editing site complimentary sequence resulted in age-dependent postnatal seizures, selective CA3 neurodegeneration at mossy fiber synapses, early death during a developmental period of high propensity to seizures (79), and increased sensitivity to Ca2+ conductance and epileptiform events (80, 81). In contrast, when a similar mutation was made in the adult brain (but was restricted only to hippocampal neurons) no neuropathological symptoms developed (82). In addition, other previously designed mutants expressing different levels of GluR2B subunit alleles that were deficient in Q/R-site editing that had increases in Ca2+ permeability did not always correlate with neurodegeneration or with the level of GluR2B (Q/R) protein expression. For example, the GluR2B editing deficient mutants (neo/neo) that had the highest Ca2+ AMPA receptor-mediated permeability were lethargic and developmentally retarded but they did not exhibit neurodegeneration or seizures suggesting very high Ca2+ levels may result in a sustained adaptation (81, 83). In contrast, the Ntrans+/+ genotype had higher levels of GluR2B protein but little or no change in Ca2+ permeability and these mice displayed late motor neuron degeneration (83). Interestingly, Seeburg and co-workers also showed that mutations of the NR1 subtype of NMDA receptors which cause or lead to low Ca2+ permeability led to impaired LTP and memory deficits but not to cell death in hippocampus or forebrain structures (84).
Does the Activation of Calcium-Permeable AMPA Receptors Lead to Cell Death?
Evidence to support a role for AMPA receptors in cell death has not been obtained, owing to the difficulty in distinguishing between glutamate-induced increases in Ca2+ levels via voltage-gated channels or activation of other subunits from AMPA receptor-mediated Ca2+ currents. Hence, in vitro studies have been carried out to examine the effects of selectively knocking down the GluR2B subunit on delayed cell death. Pharmacological increases in AMPA receptor mediated Ca2+ permeability were determined in parallel studies by optical ratio imaging with the Ca2+ dye indicator Fura 2AM (77). In vitro experiments demonstrated that exposure of GluR2B antisense deoxyoligonucleotides (AS-ODNs) to dissociated hippocampal neurons effectively produce a time-dependent reduction in GluR2B protein without inducing a generalized neurotoxicity (Figure 1A⇓, top panels). After AS-ODN–mediated knockdown of the GluR2B but not GluR1A subunit, l-glutamate- or KA-induced cell death was not enhanced despite marked decreases in GluR2B protein and increases in AMPA receptor-mediated Ca2+ permeability (Figure 1A⇓, lower panels). In fact, the opposite occurred such that a trend of reduced toxicity to l-glutamate relative to sense-treated control cultures was observed (Figure 1B⇓) and KA toxicity was unchanged (Figure 1C⇓). Accordingly, in vivo intrahippocampal infusions of GluR2B AS-ODNs into adult rats had no effect on behavior or pathology (77). In ex vivo slices prepared from seizure phenotype rat pups, LTP dropped off rapidly, suggesting that neurons may be more tolerant to glutamatergic responses—possibly by rises in Ca2+ within the cell body—that result in higher excitation of dendrites (76). Adaptation mechanisms to protect cells against the l-glutamate-mediated toxicity that arises from saturation of internal signal transduction cascades may be supported because elicitation of NMDA-dependent responses in epileptic tissue is more difficult to achieve, because of chronically elevated basal [Ca2+]i (Figure 5⇓). In addition, juvenile ages and sublethal ischemia show an opposite effect on AMPA receptor expression such that the GluR1A/GluR2B ratio decreases dramatically (45, 70) (Figure 1B⇓). These changes in GluR1A/GluR2B ratio would not lead to formation of Ca2+-permeable AMPA receptor channels and cannot be readily reconciled with the postulated damage produced by Ca2+ influx via AMPA receptors lacking the GluR2B subunit.
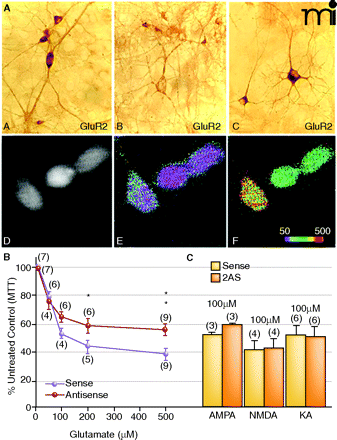
Dissociation between elevated Ca2+permeability and neurotoxicity. A. GluR1A and GluR2B immunolabeling of hippocampal neurons sevety-two hours after exposure to GluR2B AS-ODNs. (A). In sense strand–treated controls, GluR2B protein expression was intense and uniform within the somata and weaker in the dendrites of single cells. (B). After GluR2B knockdown, somatic immunolabel was highly reduced at this time. (C). In sister cultures, GluR1A protein detection was intense within the cell body as well as proximal and distal dendritic processes indicating a rise in the GluR1:2 ratio. Ca2+ imaging experiments showed corresponding pharmacological rises in AMPA receptor-mediated Ca2+ permeability. (D). Basal Ca2+ levels in GluR2B AS-ODN–treated culture. (E). Pseudocolor micrographs of basal Ca2+ levels in GluR2B AS-ODN–treated culture. (F). Puff application of AMPA under the same conditions significantly increased [Ca2+]i in the same neurons. B. and C. Effect of GluR2B knockdown on L-glutamate- (B) or KA- (C) treatment on cell viability in hippocampal cultures. On day twelve, cultures were treated with GluR2B AS-ODNs, and on days thirteen or fourteen, cultures were treated with excitotoxins. Data represent the mean ± SEM of 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) mean values at 570 nm that were quantified from control and experimental hippo-campal cultures. Modified from (77, 143). Reprinted with permission.
GluR2B AS-ODNs have been used in vivo to regulate GluR2B subunit expression unilaterally at different maturational stages in order to test directly whether a loss of GluR2B expression alters seizure susceptibility and causes cell death in the hippocampus (75–76) (Figure 2⇓). Ca2+ imaging and toxicity measurements were also performed in dissociated hippocampal cultures after GluR2B knockdown (Box 2) (77). In keeping with the results obtained from seizure phenotype mutants, unilateral hippocampal knockdown of the GluR2B but not GluR1A subunit in young rat pups in the absence of KA evoked spontaneous seizure behavior, paroxysmal activity in the EEG, and loss of long-term potentiation (LTP) in young (but not adult) rats (Figure 2⇓) (76). This result further showed that increased Ca2+ permeability is only epileptogenic during a critical stage in development.
When GluR2B antisense experiments were performed in adult rats, no cell death was observed in the absence of KA-induced seizures (75, 77). Radiolabeling of the GluR2B AS-ODNs demonstrated that they diffuse to anterior and posterior levels of the dorsal hippocampus without spreading to the ventral hippocampus or other brain regions (Figure 2A⇓). Therefore, sparing of the CA1 and dentate gyrus (DG) under these conditions was not due to lack of ODN uptake. Results from knockdown experiments were corroborated by the absence of seizures and neurodegeneration in mature GluR2B knockout mutants (85). Similarly, when seizures were induced with KA in adults after GluR2B knockdown, CA3 cell death was only enhanced in the CA3 while CA1 and DG regions were spared as was observed in the young rat pups that had succumbed to spontaneous seizures (Figure 2 B, C⇓). Thus, a GluR2B-deprived neuronal circuitry by itself does not trigger cell death but only increases the pathological consequence of the seizure, particularly at mossy fiber synapses. Together, in vitro and in vivo studies implicate a role for Ca2+ permeable AMPA receptors in “tolerance” and “adaptation” as glutamate excitotoxicity was not accentuated in other regions with increased Ca2+ permeability and neurotoxicity was reduced at high doses of glutamate after GluR2B knockdown in dissociated hippocampal neurons (77) (Box 2).
Preconditioning and Neuroprotection
Although seizures are harmful, several observations suggest that they can also protect the brain from neuronal damage. For example, in adult rats a short episode of KA-induced status epilepticus or a mild electroconvulsive shock prevents typical hippocampal injury ensued by a subsequent seizure (86–89). Similarly, audiogenic seizures reduce CA1 susceptibility to kindling in ex vivo slice preparations (90). We recently demonstrated that early-life seizures cause long-term neuroprotection of CA1 neurons during the prepubescent period (Figure 3 A, B⇓) (45). At P13, rats with either one (1×KA) or three injections of KA (3×KA) showed no histological injury of the hippocampus (Figure 1A⇓). P20 rats treated with 3×KA and a delay of seventy-two hours before sacrifice had a reduced number of CA1-injured neuronal somata; dendrites were spared (Figure 3A⇓ and Figure 4A⇓). At P30 with 3×KA, significant reductions in silver-impregnated cell bodies of the CA1 (but not of the CA3 subregion) were also observed (Figure 3D⇓ and Figure 4B⇓). Immunohistochemical analysis showed multiple early-life seizures could prevent changes in AMPA receptor expression that were induced after a single seizure. After 3×KA (on P6, P9, and P20), GluR1 and GluR2 labeling was indistinguishable from controls (Figure 3D⇓ and 4C⇓). Similarly, older animals with 3×KA (on P6, P9, and P30), showed robust AMPA receptor labeling of the CA1 region similar to control animals (Figure 3 E, F, H, J⇓ and Figure 4C⇓). Decreases in expression were only apparent in areas of the CA3 that were not spared by the earlier seizures (Figure 3D-F, H-J⇓). Quantification after 3×KA on P20 and P30 showed all hippocampal subregions had protein levels greater than 75% of control immunodensity values (Figure 4⇓). Thus, a history of early-life seizures prevented changes in the GluR1A/GluR2B ratio within protected CA1 neurons at both juvenile ages examined.
Paradoxically, intracellular Ca2+ buffering increases with maturation (44) as does the Ca2+ extrusion capacity of the Na+/Ca2+ exchanger (NCX) (91), yet seizure-induced neuronal damage only occurs after a certain age (i.e. third postnatal week). However, the ability of NCX in restoring [Ca2+]i to resting levels is decreased when glutamate is present. Therefore, when exposure to glutamate is prolonged, Ca2+ extrusion mechanisms are impaired and Ca2+ influx continues even after glutamate is cleared from the synaptic cleft (92). Because immature hippocampal neurons are resistant to damage, we hypothesized that rises in [Ca2+]i among immature neurons may be attenuated after seizures and that there would be age- and region- dependent effects of single vs multiple perinatal seizures on Ca2 influx and accumulation in the hippocampus. The FURA-2AM dye indicator was used to measure [Ca2+]i after 1×KA and 3×KA at acute and delayed time points (Figure 5⇓). Basal Ca2+ measurements were very consistent and relatively low in control slices prepared from P13 rat pups (Figure 5A⇓). In contrast to our expectation, a single episode of status epilepticus at P13 produced sustained elevations in basal Ca2+ levels in the presence of the selective Na+ channel blocker tetrodotoxin (TTX) at five hours after the KA injection such that the CA1, subiculum, and DG had the largest concentrations of Ca2+ (Figure 5A⇓). Ratiometric calculations also showed that CA2 and CA3 subregions increased their Ca2+ concentrations but to a lesser degree (Figure 5B⇓). Responses to non-saturating doses of NMDA (30 μM) were also significantly potentiated in the same subregions and these Ca2+ concentrations reversed to baseline in the presence of the selective antagonist NMDA receptor 2-amino-5-phosphonovalerate (APV). Single seizure data suggest that one episode of KA-induced status epilepticus increases hippocampal sensitivity to NMDA after seizures have subsided. On the other hand, three episodes of status epilepticus induced with 3×KA (on P6, P9, and P13) showed much smaller increases in both Ca2+ baseline (Figure 5B⇓) and NMDA stimulated responses. These data support the hypothesis that multiple perinatal seizures reduce and curtail basal elevations of [Ca2+]I and attenuate glutamate stimulated Ca2+ influx to contribute to epileptic tolerance.
Hypoxic or ischemic preconditioning models demonstrate that certain levels of [Ca2+]i can protect against subsequent insults. For example, sublethal hypoxia or glucose deprivation in organotypic (slice) hippocampal cultures produce modest elevations of [Ca2+]i (50–200 nmol) that can uncouple subsequent increases in [Ca2+]i to promote cell survival (93). Similarly, in vivo studies also show that a brief global ischemic episode induced prior to a second one of longer duration will protect CA1 hippocampal neurons from dying (94). Protective effects also have been demonstrated in focal ischemia models (94–96). Increased opening of ATP-sensitive potassium channels (KATP) were implicated in this protective mechanism in myocardial tissue (97); however, KATP channels appear not to be obligatory for ischemic preconditioning of CA1 hippocampal neurons (98). It is noteworthy that pre-exposure of β-estradiol can protect CA1 and/or CA3 neurons from cell death subsequent to seizures (99), ischemia (100), spinal cord injury (101), and in Alzheimer Disease experimental models (102). Ca2+-dependent mechanisms underlying the estrogenic effects were illustrated in cultured hippocampal neurons. For example, in vitro β-estradiol pre-treatment significantly enhanced glutamate-induced influx of Ca2+; however, stimulation of β-estradiol receptors prevented glutamate-induced excitotoxicity (103). The paradoxical neuroprotection was mediated by a modification of basal mitochondrial content of the neuron to produce a mitochondrial Ca2+ tolerance. Thus, mitochondrial sequestration of Ca2+, which attenuates excitatory-mediated increases in [Ca2+]i may be a primary mechanism of tolerance that is in part regulated by ovarian hormone-induced increases in efficiency of mitochondrial respiratory function.
Adaptive mechanisms may also be learned from non-mammalian species, such as the freshwater turtle and tadpoles, because they can tolerate prolonged periods of anoxia (104–107). Turtle neurons survive anoxia through their abilities to maintain ionic homeostasis and to limit increases in [Ca2+]i when anoxia is sustained (108). The crucian carp (Carassius carassius) is of particular interest because of its extreme anoxia tolerance and natural hypoxic preconditioning ability (109). Isolated forebrain cells from bullfrog tadpoles have elevated [Ca2+]i but not significant cell death, following prolonged anoxic conditions (110). Similarly, large increases in [Ca2+]i were not associated with neuronal death in adult frogs exposed to anoxic conditions and energy depletion (111) unless the hypoxia lasted for days (107). Painted adult turtles were also intolerant to anoxia even for months (112), suggesting that the time-scale rather than the actual magnitude of [Ca2+]i elevation is critical for neuroprotective signaling. It remains to be elucidated how large elevations of [Ca2+]i in immature neurons and moderate increases in mature neurons may result in a sustained adaptation.
Dendritic Spines: Role in Neuroprotection
Aside from the assumed role of dendritic spines in long-term memory, an important function of dendritic spines may be to protect neurons from highly elevated concentrations of Ca2+ (113). Neuroprotection may be afforded because dendritic spines of the hippocampus and cortex contain both ligand-gated (i.e., NMDA) and voltage-gated Ca2+ channels (114, 115). Hence, secondary dendritic spines have been demonstrated to independently regulate Ca2+ concentration to even higher levels than primary parent den-drites which may attenuate the toxic effects of high concentrations of Ca2+ in the neuron body by a process of sequestering the elevated concentrations of Ca2+ (116, 117). Additionally, when cultured hippocampal neurons were deprived of spontaneous ongoing network activity by chronic exposure to TTX they underwent spine loss, exhibited deficient Ca2+-clearance mechanisms, and eventually underwent neurodegeneration (118). Similarly, two weeks of TTX infusion into the rat supra-optic nucleus (SON) reduced the survival of its target neurons of the magnocellular nucleus, which was reversed by depolarization with elevated concentrations of KCl (119). Thus, the presence of a certain amount of spontaneous activity and density of dendritic spines appears necessary for the vitality of neurons at least in certain brain regions that may prevent devastating effects of neurotoxic insults.
On the other hand, blockade of spontaneous activity does not always reduce spine density because TTX exposure has time-, regional-, and age-dependent effects. For instance, although visual sensory information is required for the evolution of ocular dominance columns in the visual cortex (120), TTX application to the developing retina during axonal remodeling does not alter the normal pruning of ganglion cell spines and dendritic branching (121). Even when TTX was administered to cat fetuses during embryogenesis, retinal ganglion dendrites were similar to those observed in untreated animals, and non-beta cell dendrites increased their number of spines. This suggests spine formation within ganglion cells is independent of action potential activity in their targets and terminal branching patterns (122). Moreover, administration of TTX into the cerebellar parenchyma in adult rats generated an increase of spines on Purkinje apical dendrites and a decrease in the climbing fiber terminal arbor but with an unmodified spine density on secondary branches (123). These results indicate that there are different regional and local effects of chronic activity blockade that contribute to selective vulnerability.
Synaptic activity can increase formation of new spines by afferent stimulation (124), thus it is thus logical that elevated spine density may also occur after recurrent synchronous activity, such as in seizures. Paradoxically, intrahippocampal infusions of TTX lead to transient dendritic spine loss in the apical arborization of hippocampal CA1 pyramidal cells in adult rats after seizures were initially induced (125, 126). Interestingly, the proportion of CA1 dendritic spine-type was maintained and many spines were re-established during the latent chronic epileptic phase, when recurrent spontaneous seizures develop 3–4 weeks later (125, 126). In the immature brain, the infusion of TTX into the hippocampus during a critical epileptogenic period of CA3 network hyperexcitability (second post-natal week) (127) resulted in delayed spontaneous electrographic seizures that occurred 2–4 weeks later and continued even into adulthood (128). The reduced seizure threshold was not associated with increases in recurrent axon arbors, spine density, or additional axon sprouting, but instead with pruning of collateral neurons as a result of the ongoing epileptiform discharges (129). If TTX was introduced earlier in the neonatal period, the basal dendritic tree was only temporarily impaired and compensated by the fourth post-natal week (130). Thus, blockade of spontaneous activity has age-dependent effects on the seizure threshold, dendritic spine density, network activity, and neuronal survival. Analysis of human epileptic tissue revealed that the spine density in proximal dendrites was five times higher in dentate granule cells than in control tissue suggesting that synaptic spines of the dentate gyrus may be protected from seizure-induced excitotoxic cell damage (131). Therefore, changes in spine density by early-life seizures may reflect an adaptive response possibly by control of local Ca2+ overload as observed in our recent Ca2+ imaging study (Figure 5⇓). Tolerance was recently observed in the developing cortex such that glutamatergic transmission (effected by AMPA or NMDA treatment) can stabilize cortical spine density but stabilization was not correlated with AMPA or NMDA receptor expression (132). Therefore, activity of other receptors such as voltage-gated Ca2+ channels may be significant in regulating dendritic spine density and the amount of neural activity that is neuroprotective. In keeping with this, dantrolene, an antispastic drug that prevents Ca2+ release from intracellular stores, reduces delayed degeneration of CA1 and CA3 neurons after KA-induced status epilepticus whereas an L-type Ca2+ voltage-dependent blocker, nimodipine, does not (133). This suggests that excessive release from intracellular stores may be required for the development of delayed neurodegeneration that may be age-dependent. The effects of spine loss or gain followed by excitotoxin regimens require further investigation to determine their neuroprotective role.
Conclusion
This article has reviewed the findings that large elevations in [Ca2+]i do not always correlate with neurotoxicity but may instead have neuroprotective signaling effects. The editing incompetent GluR2B mutants, antisense knockdown approaches, and ischemia tolerance studies suggest that elevated Ca2+ can lead to reduced glutamatergic responsiveness and neuroprotection. Moreover, increases in both AMPA and/or NMDA receptor-mediated Ca2+ permeability are developmentally regulated and have a preferential contribution to normal brain function during early postnatal life.
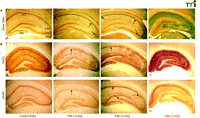
Maturational switch in neuronal vulnerability after status epilepticus. A. Photomicrographs of age-dependent kainic acid (KA) seizure-induced neurodegeneration (silver impregnation 3 days after status epilepticus). (A) KA seizures at postnatal day 13 (P13) resulted in control labeling and minimal or no argyrophilia (silver staining). (B) At P20, 1×KA produced robust injury that was preferential to the CA1 subregion (between arrows); few scattered CA3a neurons were labeled by the silver stain (arrow). (C) At P30, the CA1 was still preferentially marked by the silver stain (arrows), but many scattered neurons of the CA3a were also argyrophilic and the hilus was spared (arrow). (D) At P60, the pattern reversed such that CA3a-c and hilus were predominantly affected with high intensity labeling of dendritic arborizations (grey color); few CA1 neurons were labeled. 40× magnification. B. P20 rats: Control photomicrographs of GluR1A (A) and GluR2B (C) show uniform and intense immunolabeling of the hippocampus at 72 hrs after 1×KA, GluR1B protein is markedly decreased in CA1 and dentate gyrus (DG) but is sustained in the CA3 and ventral DG (B). GluR2B protein is decreased in CA1 (D) but much less so than that of GluR1A (B). P30 rats: GluR1A immunolabeling (E) after 1×KA shows significant depletion of protein in CA1 neurons; GluR1A was sustained in surviving CA3 neurons. In contrast, at P30 GluR2B (F) protein was increased in CA1 and marked the injured neurons. GluR2B protein was decreased in surviving CA3 subregions. (G), In contrast, at P60, GluR1A immunodensity was significantly dense in CA1 and CA3 whereas GluR2B (H) was decreased in the CA3 and hilus. Modified from (32, 45). Reprinted with permission.
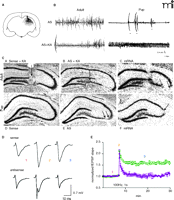
GluR2Bknockdown produces age-depenedent epileptogenicity and neurodegeneration. A.33P GluR2B antisense deoxyoligonucleotides (AS-ODN) autoradiography showed the spread and accumulation of our GluR2B antisense injection was limited to the dorsal ipsilateral hippocampus, as indicated by schematic outline of a radiolabeled coronal section. B. In adults, following knockdown of GluR2B expression in the hippocampus alone, EEG recordings showed typical asynchronous activity and normal movement artifacts (upper left trace). Similarly, GluR2B knockdown followed by KA injection did not alter the rhythmical activity in the EEG when compared to KA controls (bottom left trace). In pups, GluR2B knockdown in the absence of KA, produced spontaneous convulsive seizure-like behavior, including body tonus with fore-limb extension and laying on one side; this behavior was associated with high-frequency low-amplitude rhythmical activity of increasing size. Large movement artifacts shown represent intermittent bilateral jerking movements (asterisks) (upper right trace). In GluR2B AS-ODN–treated pups lacking the phenotypic behavior (bottom right trace), a low concentration of KA produced high-frequency rhythmical oscillations associated with tonic-clonic seizure behavior. C. As predicted by the procedure, after GluR2B knockdown and KA-induced seizures, GluR2B mRNA expression was unaltered in CA1 and DG regions but was decreased or lost only within regions of CA3 cell loss at either age [between the arrows in panel (C) and at the arrow in panel (F)]. D. There was a population spike (recruitment of firing) over the excitatory postsynaptic potential (EPSP) after GluR2B expression knockdown compared to that observed in sense-ODN–treated controls (1, baseline; 2, 100 Hz tetanic stimulation; 3, post-tetanus response). E. Long-term potentiation (LTP) was attenuated (3) after GluR2B knockdown where as the induction was unaltered (2). Modified from (75, 76, 142). Reprinted with permission.

Preservation of CA1 neurons and CA1 AMPA protein after 3×KA. A. Control photomicrographs of P20 silver stained section showing no argyrophilia. At P20 (A) and P30 (D) after 3×KA treatment, the CA1 was protected but CA3 damage shown as black staining was present. At P20 and P30, GluR1A (B, E) and GluR2B (C, F) protein levels were similar to controls (see Fig. 1⇑), except for in the CA3 region associated with cell loss (arrow). At P30, GluR1A immunolabeling (G) was significantly decreased in CA1 whereas GluR2B was increased (I). Steady levels of GluR1A and GluR2B were observed after 3×KA (H, J). Modified from (32, 45). Reprinted with permission.
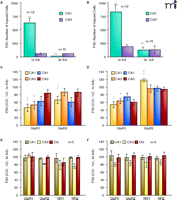
Quantification of injured and protective effects. Graph of injured cells at P20 (A) and P30 (B). AMPA receptor immunodensity shows significant decreases in GluR1A expression after 1×KA at P20 (C) and P30 (D); however, increases in GluR2B were observed at P30. AMPA and NMDA receptor immunodensities show no significant difference versus controls after 3×KA at P20 (E) and P30 (F). Bars are means ± SEM of 5–7 animals per group. One-way ANOVA, *p ≤ 0.05, **p ≤ 0.01. DGd, dentate gyrus dorsal; DGv, dentate gyrus ventral. Modified from (32, 45). Reprinted with permission.
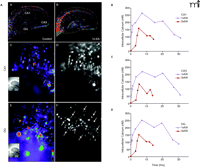
Ca2+imaging following single vs. multiple early-life seizures. A. The FURA 2AM dye indicator was used to measure intracellular Ca2+ at P13 after single (1×KA) or multiple (3×KA) episodes of KA-induced seizures that were induced on P6, P9, and P13. (A). Ca2+ visualization of hippocampal neurons loaded with Fura-2AM in control slices show low amounts of Ca2+ in presence of TTX. (B). Five hours after 1×KA treatment, basal Ca2+ amounts were elevated throughout the hippocampus but particularly in CA1 [(C), (D)] and DG [(E), (F)]. B. Time course of elevated basal Ca2+ concentrations after 1×KA vs 3×KA in CA1 (B), CA3 (C), and DG (D).
Acknowledgments
The research was supported by the NIH National Neurological Disorders and Stroke NS-38069 and Partnership for Pediatric Epilepsy, 589045.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Linda K. Friedman, PhD, is an Associate Professor of Neuroscience at the New York College of Osteopathic Medicine of the New York Institute of Technology (NYIT). She received her doctoral degree from the Department of Neuropsychology, City University of New York Graduate Center where she trained under Dr. Mytilineaou from the Department of Neurology at the Mt. Sinai School of Medicine. Dr. Friedman’s research explores mechanisms that are responsible for the susceptibility of specific neuronal populations to excitotoxic damage as a function of age and seizure history. E-mail: lfriedma{at}nyit.edu, fax 516-686-7950.

