Twenty Years of Calcium Imaging: Cell Physiology to Dye For
- Harm J. Knot1,
- Ismail Laher2,
- Eric A. Sobie3,
- Silvia Guatimosim3,
- Leticia Gomez-Viquez3,
- Hali Hartmann3,
- Long-Sheng Song3,
- W.J. Lederer3,
- Wolfgang F. Graier4,
- Roland Malli4,
- Maud Frieden5 and
- Ole H. Petersen6
- 1Department of Pharmacology & Therapeutics and Division of Cardiology College of Medicine, University of Florida, Gainesville, FL
- 2Department of Pharmacology and Therepeutics, University of British Columbia, Vancouver, BC
- 3Medical Biotechnology Center, University of Maryland Biotechnology Institute, Baltimore, MD
- 4Institue of Molecular Biology & Biochemistry, Medical University of Graz, Austria
- 5Department of Cell Physiology and Metabolism, University of Geneva Medical Center, Geneva, Switzerland
- 6The Physiological Laboratory, University of Liverpool, Liverpool, UK
Abstract
The use of fluorescent dyes over the past two decades has led to a revolution in our understanding of calcium signaling. Given the ubiquitous role of Ca2+ in signal transduction at the most fundamental levels of molecular, cellular, and organismal biology, it has been challenging to understand how the specificity and versatility of Ca2+ signaling is accomplished. In excitable cells, the coordination of changing Ca2+ concentrations at global (cellular) and well-defined subcellular spaces through the course of membrane depolarization can now be conceptualized in the context of disease processes such as cardiac arrhythmogenesis. The spatial and temporal dimensions of Ca2+ signaling are similarly important in non-excitable cells, such as endothelial and epithelial cells, to regulate multiple signaling pathways that participate in organ homeostasis as well as cellular organization and essential secretory processes.
Introduction
Since the first description and use of fluorescent dyes for measurement of intracellular free calcium concentration ([Ca2+]i) twenty years ago (1), biological scientists have gained unprecedented insights into the mechanisms of cell signaling. In particular, the use of fluorescence spectroscopy has made possible the detection of the minute levels of [Ca2+]i that exist in relative obscurity compared to the large reservoir of bound or sequestered Ca2+ in stores within most cells. Although allusions to the physiological importance of calcium certainly was recognized earlier (Box 1), the role of Ca2+ as a communicator and regulator of cell function is entrenched in the literature primarily through studies using fluorescent dyes, in various cell types, reported over the past two decades. We now appreciate the complexity arising from distinct—often simultaneous—Ca2+ signaling entities across the temporal and spatial domains of many cells, as well as multiple entities occurring within a single cell type (Table 1⇓).
Ca2+ Signaling Entities in the Vascular Smooth Muscle Cell
Recognition of Calcium Signaling.
“Ja Kalzium, das ist alles!” (Yes, calcium, that’s everything!)
—O. Lowei (1959)
“A small ion with cachet...that old signal crackerjack...”
—A. Loewenstein (2).
Of course, there are critically important roles for other ions in biological events, such as the Na+-dependent action potentials underlying nerve and muscle function, but why has evolutionary biology favored Ca2+ above all other ions as a universal signal? Owing to its unique physicochemical properties, cells have evolved a number of proteins (e.g., calmodulin and troponin C) that can bind Ca2+ with high affinity at physiological pH. Specifically, these binding proteins are rich in glutamate and aspartate residues. When only two of these amino acid residues are appropriately positioned within 10nm of each other, they can create a low affinity-binding (millimolar) site for Ca2+ (and Mg2+). The addition of only one or two carboxylic groups further to this constellation can confer high affinity–binding (micromolar) and a marked preference for Ca2+ over other ions.
The very steep concentration gradient for Ca2+ across the plasma membrane creates ideal conditions for using Ca2+ as a messenger. The resting [Ca2+]i ranges from 30–150nM; the extracellular Ca2+ concentration is about 10,000 times greater, which favors Ca2+ entry into cells, so that even short bursts of Ca2+ entry will generate relatively large signals. Thus, during activation, the [Ca2+]i can increase to 5μM or more—with concentrations in restricted spaces and intracellular compartments reaching considerably higher levels (to 100μM). The net effect of these findings is that the Ca2+ signal exceeds the resting level by up to 100-fold, setting a signal-to-noise ratio that can reach 10,000. Compared to the movement of Ca2+ in simple buffer solutions, the diffusion of the ion under physiological conditions is much slower (13–65μm2/sec), due largely to the presence of intracellular buffers.
The current family of fluorescent dyes for Ca2+ imaging was initially based on a bis(2-aminophenoxy)ethane tetraacetic acid (BAPTA), and subsequently on a bis(2-aminoethyl ether) tetraacetic acid (EGTA), backbone conjugated to a fluorescent moiety. Fura-2 is the most widely used fluorescent dye for monitoring intracellular Ca2+; it is relatively easy to load into cells in its acetoxymethyl ester form, allows for ratiometric spectroscopy [see (3)], and is relatively photostable. In solutions that mimic the cytoplasm, the Kd for the fura-2·Ca2+ complex is 224nM at 37°C, so that it is useful for detecting changes of Ca2+ concentration that occur under physiological conditions.
Several fundamental features of Ca2+ signaling have become apparent through fluorescent imaging—among them, notions such as a global (referring to arterial wall tissue) versus compartmental or local (subcellular) increases in intracellular Ca2+. This notion has profound implications; for example, spontaneous increases in local Ca2+ concentration have contrasting effects—contraction in cardiac muscle and relaxation in smooth muscle. Such observations lead to the intriguing realization that, in addition to changes in [Ca2+]i, the physical arrangements of ion channels, pumps, exchangers, and organelles play an important role in shaping the amplitude of the Ca2+ signal. These arrangements, furthermore, determine the specialized nature of the biological response; for example, global increases in Ca2+ concentration lead to constriction (myogenic tone), whereas localized, targeted increases of [Ca2+]i, termed “Ca2+-sparks” (Figure 1⇓), promote relaxation of arteries (4). In endothelial cells, such an arrangement may also explain how the same ion (Ca2+) is required for the release of both vasodilator and vasoconstrictor substances. Another aspect of cell function that has been unraveled through the use of Ca2+ indicator dyes is the detection of oscillations in Ca2+concentrations during cell activation, which revises the previous concept of amplitude-based Ca2+ signaling to one that includes frequency modulation (5). [An excellent interactive review of the role of calcium signaling can be found on the Web (6).]
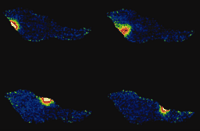
Calcium sparks. These signals are spontaneous, transient, small bursts of sub-membrane Ca2+ originating from clusters of Ca2+-release channels on the sarcoplasmic reticulum (Ca2+ stores) as shown here for an isolated vascular smooth muscle cell at 4 time points (>500 ms apart).
We have also learned much of the central role for Ca2+ in other biological events that are quite diverse in nature. The leading edge of pseudopodia during the movement of unicellular organisms has gradients of increases in [Ca2+]i that are related to directionality. Additional roles for Ca2+ include cell motility, proliferation, differentiation, and apoptosis. Through the use of dyes such as Fura-2, we have a greater appreciation (quantitative and qualitative) for the rise and fall of Ca2+ concentrations during each heartbeat, in addition to the startling detection of Ca2+ sparks in various cell types.
Although many elements of excitation-contraction coupling were previously well defined, the advent of quantitative fluorescence techniques has advanced our understanding of novel pathways of cell regulation (Table 1⇑). The use of fluorescent indicator dyes confirms that in most cells, agonists generate a biphasic signal comprised of an initial burst of Ca2+ release followed by a more sustained phase of Ca2+ entry from the extracellular space in a process termed “capacitative Ca2+ entry” (7). An example of the complexity and multidimensional nature of intracellular Ca2+ signaling that has been revealed through the use of fluorescent dyes is indicated in Figure 2⇓.
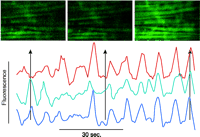
Confocal image of a rat mesenteric small artery loaded with a Ca2+-sensitive dye. The horizontal streaks are smooth muscle cells with an overlaying network of sympathetic nerve fibers. The graph shows a time series of three smooth muscle cells. The Ca2+ oscillations are first unsynchronized and later synchronized. The scale bar is 4 μm. (Image kindly provided by Christian Aalkjaer, University of Aarhus, Denmark.)
With the availability of fluorescent indicators, it is now quite feasible to monitor proteins such as kinases, phospatases, and cytoskeletal proteins; to track nucleic acid movement; to monitor dynamically multiple intracellular ions (e.g., Na+, Mg2+, Zn2+), pH, and membrane potential; and to investigate the roles of signaling molecules such as nitric oxide and inositides. It is also possible to use fluorescent probes of various intracellular organelles, neurotransmitter receptors, ion channels, and carriers. We now have available a variety of “caged” compounds (i.e., compounds chelated by photolyzable indicators) that allow us to image not only Ca2+, but also ATP, various neurotransmitters, and other signaling molecules. The list of fluorescent dyes and their application has grown extensively and in so doing reveals our new appreciation of cell organization and regulation.
Spatial Heterogeneity of Ca2+ Signaling in Heart: A Subcellular Arrhythmic Mechanism
Excitation-contraction Coupling and Ca2+-induced Ca2+ Release
Normal heart function involves complex bidirectional interactions between electrical and chemical signaling systems within cardiac muscle cells (myocytes). These interactions must occur synchronously to ensure a strong heartbeat and efficient pumping of blood and must remain well-controlled to ensure stability. When the normal pathways for interaction break down, as can occur in disease states, uncontrolled and/or asynchronous behavior can occur. Here we briefly discuss a particular mechanism through which defective calcium signaling in heart cells can lead to a disruption in the heart’s electrical signaling known as an arrhythmia.
Several inter- and intracellular signaling steps must occur in a synchronous and controlled manner each time the heart beats. An electrical signal, known as an action potential, originates in the sinoatrial node and propagates to each cell in the myocardium. This results in the membrane potential (Vm) in each ventricular cell changing from a resting value of ~−80 mV to a peak value of around +40 mV. This membrane depolarization underlies a large increase in intracellular Ca2+ concentration ([Ca2+]i), and cellular contraction results from Ca2+ ions binding to myofilaments. The increase in [Ca2+]i, known as a Ca2+ transient, is therefore the chemical signal that transduces electrical depolarization to mechanical movement, a sequence of events collectively known as excitation-contraction (EC) coupling. Experiments performed nearly 20 years ago (8) established that this key step in EC coupling occurs because a small influx of trigger Ca2+, primarily that flowing through L-type Ca2+ channels (9), causes the release of a much larger amount of Ca2+ from the sarcoplasmic reticulum (SR). This process, known as Ca2+-induced Ca2+ release (CICR) therefore displays both high gain and positive feedback, characteristics that in principle can lead to regenerative, all-or-none behavior and instability. However, in healthy hearts, CICR is well controlled, and Ca2+ release is a graded rather than an all-or-none phenomenon (10). This paradox of a graded, high-gain, positive feedback system inhibited the development of a mechanistic understanding of CICR for many years.
Local Control of CICR and Ca2+ Sparks
The solution to this paradox is the fact that Ca2+ release is under “local control.” According to this idea (11, 12), because current flowing through an open L-type channel will raise Ca2+ concentration to a significant extent only in the immediate vicinity of the channel (13), Ca2+ entry can trigger release from SR release channels (known as ryanodine receptors, RyRs) located nearby (i.e., within 100 nm), but will have no effect on more distant RyRs. Direct evidence of the local control hypothesis came with the discovery of Ca2+ sparks (14–16) or localized increases in [Ca2+]i caused by the opening of one or a cluster of RyRs. Subsequent studies have demonstrated that the graded nature of CICR stems from the recruitment of many Ca2+ sparks (17), even though each individual Ca2+ spark is an all-or-none event, and that ventricular cells isolated from failing hearts can display an impaired ability to trigger Ca2+ sparks (18). Thus, investigations of the properties of Ca2+ sparks provide critical information about how cardiac CICR is regulated in health and disease.
Misregulation of Cardiac CICR in Disease
Normally, local control of CICR allows the cell to keep this process in check, but under pathological conditions Ca2+ release can become unstable such that a spontaneous Ca2+ spark will in turn trigger sparks from neighboring sites, and a propagating, regenerative Ca2+ wave will result. Experiments have demonstrated that waves generally occur under conditions of Ca2+ overload (19, 20); Ca2+ waves induce inward current as the increased cytosolic Ca2+ is extruded from the cell via the Na+–Ca2+ exchanger because three Na+ ions enter the cell for every Ca2+ ion that exits (21, 22). The inward current that results from spontaneous intracellular Ca2+ release causes the membrane potential to become more positive, known as a delayed afterdepolarization (DAD; “delayed” because they often occur during diastole). A DAD can cause an ectopic action potential that can in turn lead to an arrhythmia; indeed, Ca2+-linked arrhythmias arising from ectopic beats are thought to be the main cause of sudden cardiac death in heart failure (HF) (21, 23). Paradoxically, however, changes in Ca2+ cycling proteins that occur in HF, such as enhanced Na+–Ca2+ exchanger and depressed sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) function (24, 25), tend to decrease SR Ca2+ load. Reduced load in HF would be expected to result in increased rather than decreased CICR stability. Thus, the mechanisms that underlie the increased Ca2+ instability and increased risk of arrhythmia in HF remain largely a mystery, and many investigations on the pathophysiology of HF are therefore focused on uncovering these mechanisms.
Ca2+ Overload, EADs and DADs
One circumstance that has been known to promote instability and arrhythmias for over thirty years is “Ca2+ overload,” the condition in which cardiac myocytes have an excessive amount of Ca2+ (26). “Early afterdepolarizations,” or EADs, abnormal depolarizations that occur during the repolarizing phase of the cardiac AP and are due to interactions between the depolarizing Ca2+ current and repolarizing potassium currents (27), are enhanced by Ca2+ overload (28). DADs, which by definition develop after complete repolarization of the membrane, also tend to occur under conditions of Ca2+ overload (29). The idea that oscillatory Ca2+ release is one of the major factors underlying these Ca2+-dependent arrhythmias was appreciated early in the investigations of Ca2+ overload and arrhythmogenesis (30), and spatial heterogeneity of the Ca2+ instability was suspected by studies of the current noise that developed during Ca2+ overload (30–33). Although complex diffraction patterns were observed when the cell and tissue were viewed with a laser (34), they were not characterized until relatively high-speed imaging of single cardiac myocytes was possible (35). In these and related experiments a global or cell-wide increase in the total amount of Ca2+ within the cells led to an overall elevation of the SR “Ca2+ load.” In addition, in these early studies propagating waves of elevated Ca2+ were observed initiating from various sites. More direct observations of the source of the local [Ca2+]i increases, and more mechanistic insight into the causes of Ca2+ waves, were possible with the identification and characterization of Ca2+ sparks (16).
Normal Ca2+ Sparks
Ca2+ sparks occur at transverse tubules (TTs) (36), as shown in Figure 3⇓. In quiescent rat ventricular myocytes sparks occur at a “low” rate of around 100 per cell per second (16) but can be synchronized to produce a normal [Ca2+]i transient by the opening of voltage-gated L-type Ca2+ channels or dihydropyridine receptors (DHPRs) upon membrane depolarization (18, 37, 38). More recently, it has become apparent that many DHPRs may be activated to redundantly trigger a Ca2+ spark in rabbit ventricular myocytes (39). Thus, there are species differences in the organization of the DHPRs that trigger Ca2+ sparks, as well as in the SR Ca2+ load (30) and number of RyRs that may form a cluster in the SR (40). Nevertheless, there is significant stability in EC coupling and little, if any, Ca2+ -dependent arrhythmogenesis under control conditions in normal ventricular myocytes.
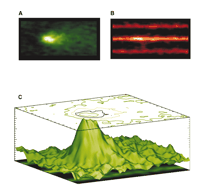
Calcium sparks and transverse tubules (TTs). The Ca2+ indicator fluo-3 was loaded into heart cells to enable Ca2+ sparks to be visualized on a confocal microscope. A. Signal-averaged Ca2+ sparks. B. Sulforhodamine B was added to the extracellular solution to image the TTs, and Ca2+ sparks were imaged simultaneously. C. A surface plot shows the relationship between signal-averaged Ca2+ sparks (A) and TTs (B). The site of the origin of the majority of Ca2+ sparks is the TT from the junctional SR (jSR). [See (29).]
Ca2+ Overload
When SR Ca2+ content increases, Ca2+ sparks occur at a higher rate (15, 41). In addition, Ca2+ sparks can trigger Ca2+ waves (15), and these may activate the Na+–Ca2+ exchanger, which can trigger extrasystoles (15, 21). This arrhythmogenic mechanism was basically outlined in the early investigations of Ca2+ overload (26, 28–30) and has been refined to include local signaling. However, this leads to a paradox, as mentioned above, because heart failure of diverse causes leads to both a decrease in global SR Ca2+ load (18) and Ca2+-dependent arrhythmogenesis. How can these arrhythmias occur in the context of a reduction of global averaged Ca2+ content? Our approach to this paradox considers that two additional changes may be observed in cardiac myocytes in addition to the decrease in global SR load: 1) spatial heterogeneity in SR Ca2+ content —in particular, the observation that some junctional SR (jSR) elements are overloaded while some jSR are “under-loaded;” and 2) increased sensitivity of RyRs to trigger Ca2+ (Figure 4⇓). These two changes together may be sufficient in principle to enable a Ca2+ spark to activate a Ca2+ wave despite the cell-wide decrease in SR Ca2+ content (Figure 5⇓). The propagating wave may be sufficient to produce a DAD or augment the probability of an EAD without additional cellular changes. EADs and DADs can give rise to extra-systoles and thus may underlie an arrhythmia. If expression of the Na+–Ca2+ exchanger is increased and/or if repolarizing K+ currents are decreased—conditions frequently characteristic of HF—then the likelihood of EADs and DADs producing extrasystoles and arrhythmias is increased still further (21).
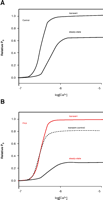
Sensitivity of RyR2s and Ca2+triggering. A. Transient elevations of [Ca2+]i are better able to activate RyR2s than steady-state changes. Here, the RyR2 open probability (Po) increases more following a step increase in [Ca2+]i than it does in the steady state for the same final [Ca2+]i. B. PKA enhances the relative increase in Po due to a step increase of [Ca2+]i but decreases the steady-state Po.

Effects of spatial heterogeneity of SR Ca2+content. A. Diagram of the cross-section of a cell with the SR Ca2+ release sites (or jSR or Ca2+ release “units”) represented as open circles. B. Time sequence (i–iv) of [Ca2+]i images of control heart cell with normal Ca2+ load in the jSR represented as black filled circles. Ca2+ sparks occur at a low rate and do not trigger other Ca2+ sparks or Ca2+ waves. C. Time sequence of [Ca2+]i images of a cell with global Ca2+ overload. The jSR sites with elevated Ca2+ content are represented as red filled circles. Ca2+ sparks can trigger neighboring jSR to produce a propagating wave of elevated [Ca2+]i. D. Time sequence of [Ca2+]i images of a cell with heterogeneously elevated Ca2+ in SR (red filled circles) mixed with jSR regions with normal or low Ca2+ content (black filled circles). Within contiguous regions of elevated SR Ca2+, aborted Ca2+ waves may occur but propagated waves do not normally traverse the cell. E. The same jSR Ca2+ load heterogeneity as in panel D but with increased sensitivity of the RyRs (See text). Under these conditions, a Ca2+ spark can initiate a Ca2+ wave.
Heterogeneity of SR Ca2+ Content
The heterogeneity of SR Ca2+ content, a key element in our approach to understanding the paradoxical changes of Ca2+ levels in HF, can occur due to structural changes associated with pressure overload HF (21) or HF following myocardial infarction (18). Functional heterogeneity could also occur in the context of poor triggering of DHPRs, which could occur in conditions that may lead to alternans (42) or following the use of Ca2+ or Na+ channel blockers or other agents that may lead to EC coupling triggering dispersion. In each case, the heterogeneity of SR Ca2+ content comes about because a region was not triggered normally to release its Ca2+ content. This triggering dispersion can be modeled and leads to some simple features in the [Ca2+]i signals that can be readily measured. Triggering failure can lead to “dyssynchrony” (43). It will also retard the apparent “rate of rise” of the [Ca2+]i transient. Additionally, there will be more “noise” or a greater variance in the [Ca2+]i signal. These features are presented in a model shown in Figure 6⇓.
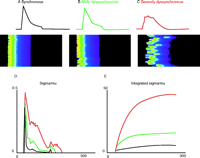
Variance and dyssynchrony. The conditions of SR Ca2+ load heterogeneity (and other conditions of triggering heterogeneity) can lead to dyssynchronous activation of Ca2+ sparks. Here, a mathematical model reveals how increasingly dyssynchronous SR Ca2+ release may affect the [Ca2+]i transient and fluctuations of [Ca2+]i. A. Nearly synchronous Ca2+ release and a [Ca2+]i transient over the period of one second. B. Mildly dyssynchronous Ca2+ spark activation (same number of Ca2+ sparks) slows the time-to-peak [Ca2+]i transient, decreases the peak [Ca2+]i level, and is associated with the development of [Ca2+]i noise. C. Severely dyssynchronous SR Ca2+ release produces an even slower rise and smaller peak of the [Ca2+]i transient and this is accompanied by even more [Ca2+]i signaling “noise.” The same number of Ca2+ sparks is activated in panels A, B, and C. D. The time course of the standard deviation is plotted as a function of time (ms) normalized by the mean [Ca2+]i. E. The integral of the signals in D.
Regulation of Endothelial Cell Function: Versatility and Specificity of Ca2+ Signaling
In comparison to excitable cells, for which the importance of Ca2+ in cell function had already attracted attention for some time, Ca2+ signaling in endothelial cells failed to receive recognition until the late 1980s. Regard for the wider versatility of Ca2+ signaling was made possible by Roger Tsien’s landmark developments in fluorescent techniques to determine intracellular Ca2+ concentration ([Ca2+]i), particularly with the introduction of the now long-time star, fura-2 (1). More recent developments in the field include targeted fluorescent protein–based Ca2+ sensors such as cameleons (44) and pericam (45). Parallel to the revolution in Ca2+ signaling (46–49), endothelial cell biology was witnessing the thrilling discovery of the so-called endothelium-derived relaxing factor and its identification as nitric oxide (50). Soon thereafter, the regulation of nitric oxide production by Ca2+ became evident (46, 51–53).
During the last decade, novel endothelial functions were being continually discovered, and the endothelium metamorphosed conceptually from a passive barrier between tissue and blood to its current status as a highly resourceful, multifunctioning and integrating organ. Remarkably, in nearly every established signaling pathway identified in endothelial cell fate and function, Ca2+ was found to play a role. In other words, in our recent concept of the endothelial “reactome,” Ca2+ is incorporated as an initiator or modulator of a vast number of signaling phenomena (see http://www.reactome.org/). Calcium’s roles are in many respects pivotal to cell function, such as its action in the regulation of post-translational protein processing and folding (54).
Given its seemingly contradictory roles in such processes as the generation of the vasorelaxant nitric oxide as well as that of the vaso-contracting endothelins (55), we need to ask how the selectivity and specificity of Ca2+ is achieved in any given type of cell. Sir Michael Berridge introduced the landmark concept that amplitude (AM) and frequency (FM) modulate Ca2+ signaling (56). Based on this concept, the following discussion briefly explores Ca2+ versatility for proper endothelial cell function.
Amplitude of [Ca2+] Elevation
The most obvious way to discriminate between two Ca2+ signals might be the amplitude of [Ca2+] elevation, so that subsequent Ca2+-sensitive downstream signaling would respond in a concentration- dependent manner to elevated [Ca2+]; however, this concept fails to address the necessary specificity in terms of downstream sensing of the Ca2+ message and merely reflects a quantitative correlation (between the extent of [Ca2+] elevation and its effect on downstream signaling). Such a correlation can account for one given agonist over a wide range of concentrations, but the model is not satisfying once one intends to compare the efficiency of two different stimuli in signal transduction. To expand upon the concept, therefore, we can apply a threshold phenomenon for distinct pathways that are only initiated after a certain [Ca2+] threshold has been reached. Such a mechanism would exhibit a very steep concentration response correlation, and in fact occurs, at least according our recent knowledge, very rarely.
Temporary Patterns of the Ca2+ Signaling
A striking property of cellular Ca2+ signaling is that, depending on the stimulus applied or even the concentration of a given agonist, the kinetics of Ca2+ signaling exhibit various patterns.
Duration
Stimulation of most seven-helix receptors that are coupled with phospholipase Cβ via a G protein (e.g., bradykinin and histamine) induces an instant elevation in cytoplasmic [Ca2+]i with a subsequent long-lasting plateau phase. Notably, two distinct patterns of seven-helix receptor–initiated Ca2+ signaling are reported in porcine coronary endothelial cells, where bradykinin initiates long-lasting [Ca2+] elevation, whereas in response to substance P, cytosolic [Ca2+] only transiently increases (57). Similar results are obtained in calf pulmonary endothelial cells, where bradykinin again evokes a long-lasting [Ca2+] rise, whereas the effect of anandamide is more transient, despite the fact that Ca2+ entry in the classical Ca2+ re-entry protocol (46) is almost identical for both agonists (58). Considering the differences in the duration of cytosolic [Ca2+] elevation, differences in the activation patterns of Ca2+-sensitive signal transduction pathways seem reasonable.
Ca2+ Kinetics
Besides the specific duration-linked stimulation of Ca2+-activated pathways, the different kinetics of [Ca2+] elevation has been reported to trigger certain pathways in endothelial cells. Notably, these cells often exhibit oscillatory Ca2+ signaling that depends on intracellular [Ca2+] homeostasis and quantal Ca2+ entry (49, 59–62). Frequency-modulated signaling has been reported for transcription factors, such as NFκB (63) and CREB (64), and major signaling pathways such as JNK (65). Remarkably, the kinetic patterns of endothelial cell Ca2+ signaling even with only one agonist are not identical and often exhibit a transient, oscillatory, or long-lasting response at low, moderate, and high agonist concentrations, respectively. Considering such concentration-dependent variation in the kinetics of [Ca2+] elevation, one might expect that the regulation of Ca2+-sensitive processes by one agonist offers variability in the force of stimulation that is elicited not merely by the quantity, but also the frequency of cytoplasmic [Ca2+] elevation.
Spatial Ca2+ Gradients
Recently, the story of the regulation of Ca2+-sensitive signal transduction in endothelial cells opened another important chapter: Localized Ca2+ signaling. Based on an existing concept in smooth muscle cells (66), for example, the appearance of localized Ca2+ gradients was hypothesized in endothelial cells and found to be established between superficial domains of the endoplasmatic reticulum and the plasma membrane [i.e., subplasmalemmal Ca2+ Control Unit (SCCU)] (67) and the mitochondria-plasma membrane junction [i.e., mitochondrial buffer unit (MBU)] (68). So far, the SCCU, which promotes high local [Ca2+] elevation in endothelial cells, has been described in the local activation of Ca2+-activated K+ channels (67) and direct Ca2+ refilling of the endoplasmic reticulum (68). In contrast, the MBU buffers subplasmalemmal [Ca2+] to basal levels even if cytosplsmic [Ca2+] is very high and thus facilitates maintenance of the Ca2+ entry activity through the Ca2+-inhibitable capacitative Ca2+ pathway (69, 70). Significantly, the mitochondrial integrity is subject to substantial changes under pathological conditions (e.g., hyperglycemic conditions) (71). Because changes in the structural organization of mitochondria affect organelle and cellular Ca2+ homeostasis (71, 72), it is tempting to speculate that disturbances in the establishment of spatial [Ca2+] gradients are causally involved in endothelial dysfunction under pathological conditions.
Distinct Effector Distribution
In association with locally isolated Ca2+ gradients, Ca2+-sensitive effector proteins may distinctly accomplish selectivity of Ca2+-triggered signal transduction. This emerging topic still needs to be explored in much more detail, but there is evidence for the differential distribution of Ca2+-sensitive proteins of certain pathways within one given type of cell. Although there is a vast amount of such Ca2+-sensitive effectors with distinct subcellular distributions (e.g., enzymes and transcription factors), the correlation of protein activation and local Ca2+ signaling has been only occasoually investigated. For example, the CBP/CREB proteins are found to be activated (i.e., phosphorylated) by Ca2+ of different compartments (e.g., cytosol vs nuclear) and even cytosolic domains (e.g., subplasmalemmal vs deeper cytosol) (73, 74). In this way, endothelial nitric oxide synthase may be subject to spatial Ca2+ signaling, because this enzyme depends on Ca2+ influx rather than intracellular Ca2+ release (46). Moreover, it has been shown that nitric oxide synthase is also localized within the mitochondrial matrix, thus limited to a different Ca2+ microenvironment from the cytosolic/plasma membrane enzyme (75).
Spatial Recruitment of Scaffold Proteins
A very interesting and straightforward premise for specific signal initialization based on individual protein distribution and spatial Ca2+ signaling could be that pathway-building scaffold proteins are distinctively distributed within the cell and are recruited by spatial Ca2+ elevation. Consequently, a spatial Ca2+ signal may activate a certain pathway by local recruitment of the respective scaffold protein. Ricardo Dolmetsch and coworkers have reported that the L-type Ca2+ channel works also as a Ca2+-activated scaffold protein for Ca2+/calmodulin in mitogenic signal transduction (63). Based on these intriguing and exciting data, one can speculate that spatial Ca2+ signaling might recruit distinct Ca2+-sensitive scaffold proteins (e.g., AKAP79 or β-arrestins) (76, 77) and subsequently trigger Ca2+-activated signal transduction selectively.
Feedbacks Loops and Priming by Ca2+
The participation of (spatial) Ca2+ signaling in localized signal transduction might be further accomplished by associated signals that are initiated independently of the Ca2+ signal—or even upstream to it—and affect the subsequent process of protein stimulation by Ca2+. A prominent example of such a possibility is vascular endothelial growth factor, which triggers intracellular Ca2+ mobilization via phospholipase Cγ (78) and requires an upstream (tyrosine) kinase pathway (79) that might secondarily modulate the role of Ca2+ in promoting signal transduction. Thus, in contrast to a seven-helix receptor–G protein–phospholipase Cβ initiated signaling that might occur as a single independent phenomenon, phospholipase Cγ-mediated intracellular Ca2+ mobilization is associated with the activation of other signaling pathways that may modulate the potential of Ca2+ as second messenger. A similar situation might exist for capacitative Ca2+ entry, a phenomenon that takes place subsequent to Ca2+ depletion of the endoplasmic reticulum and that might otherwise modulate the potential of entering Ca2+ to initiate signals. In extension of such considerations, one needs to consider that cells can be primed by various physiological and pathological stimuli that result in changes in the cellular integration of [Ca2+] elevation into meaningful signals (80). The versatility of Ca2+ signals is indicated in Figure 7⇓.
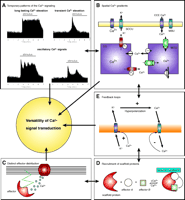
Putative mechanisms that might accomplish the versatility of Ca2+signal transduction. A. Quantitative and kinetic determination of the Ca2+ message by amplitude and frequency of global/cytosolic Ca2+ elevation. B. By generation of locally isolated Ca2+ gradients, Ca2+ selectively affects Ca2+-sensitive proteins. The schema illustrates the appearance of such areas with high (i.e., subplasmalemmal Ca2+ Control Unit; SCCU) and low (mitochondrial buffer unit; MBU) Ca2+ concentration between the plasma membrane and the respective organelle. C. By distribution of Ca2+-sensitive proteins to areas of distinct Ca2+ regulation, specificity of Ca2+-initiated signal transduction can be achieved. D. A local Ca2+ elevation recruits a Ca2+-activated scaffold protein that assembles a certain signaling pathway independently of global Ca2+ concentration. E. Ca2+ exhibits auto-regulatory properties and modulates subsequent Ca 2+ signaling.
Ca2+-Mediated Regulation of Cell Secretion
Katz and Miledi (81) and Baker and Knight (82) demonstrated convincingly the importance of Ca2+ for the control of exocytotic secretion. Here, we focus on the pancreatic acinar cell, the classic cell biological model for studies of the intracellular secretory pathway (83). The highly polarized structure and function of this cell, with protein synthesis at the base, vectorial processing of the secretory products in the basal–apical direction, and exocytotic secretion confined to the apical surface cell membrane, offers considerable advantages for studies of Ca2+ signaling. Furthermore, the acinar cells also secrete fluid, which is important for the transport of secreted digestive enzymes into the gut (84). Thus, it is possible in this cell type to study Ca2+ regulation of both exocytosis and fluid secretion.
The exocrine acinar cells, like most epithelial cells, lack voltage- gated Ca2+ channels, and depolarization therefore does not elicit Ca2+ signals or secretion (85). On the other hand, these major protein- synthesizing cells possess an extensive endoplasmic reticulum (ER) (83), and studies of exocrine acinar cells first demonstrated neurotransmitter- and hormone-elicited release of Ca2+ from the ER (85, 86). Ten years later, it was again work on pancreatic acinar cells that led to the discovery of inositol 1,4,5-trisphosphate (IP3) as an intracellular messenger liberating Ca2+ stored in the ER (87). The discovery and description, 20 years ago, of a new generation of Ca2+ indicators with greatly improved fluorescence properties (1), made Ca2+ imaging possible and has had enormous consequences also for studies of secretory cells.
Overall Ca2+ Homeostasis
Early work on perfused submandibular glands (86) established that acetylcholine (ACh) or adrenaline releases Ca2+ from a non-mitochondrial intracellular store, most likely the ER, and that thereafter Ca2+ pumps in the plasma membrane extrude the released Ca2+. The delayed uptake of Ca2+ from extracellular solution, later identified as store-operated Ca2+ entry, was also described. Studies of the external Ca2+ dependence of enzyme secretion showed that the initial secretory response to ACh was completely independent of the presence of Ca2+ in the external solution; however, a later sustained phase of ACh-induced secretion was shown to be acutely dependent on external Ca2+ (88).
The simplest model of Ca2+ transport requires information about the Ca2+ concentrations in the ER ([Ca2+]ER), the cytosol ([Ca2+]i), and the extracellular solution. The droplet technique (89) allows simultaneous measurements of [Ca2+]i as well as the [Ca2+] in a small droplet of extracellular solution surrounding an isolated cell. In later studies, by Mogami et al. (90), it was possible to measure simultaneously [Ca2+]i and [Ca2+]ER. From these studies, an overall model of Ca2+ homeostasis in pancreatic acinar cells emerged (Figure 8⇓). Maximal ACh stimulation liberates all Ca2+ stored in the ER, via opening of Ca2+ release channels in the ER membrane, and this Ca2+ is then extruded into the external solution by the plasma membrane Ca2+ pump. Thus, after several minutes of continuous stimulation, all Ca2+ stored in the ER would be exported from the cell. Cessation of ACh stimulation closes the Ca2+ release channels in the ER, allowing the ER Ca2+ pumps to refill the ER with Ca2+. Most of the Ca2+ required for ER Ca2+ uptake comes from outside the cell and enters through store-operated Ca2+ channels.
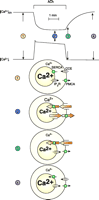
Overall Ca2+homeostasis in pancreatic acinar cell. The top two traces show [Ca2+] in the lumen of the ER and the cytosol, before, during, and after maximal ACh stimulation. Below are shown representations of the state of the four principal Ca2+ transporters in four situations: 1) Rest, before start of stimulation. Small fluxes of Ca2+ through Ca2+ release channels in the ER (for simplicity only the IP3 receptor is shown), ER Ca2+ pumps (SERCA), the plasma membrane Ca2+ pump (PMCA), and the store-operated Ca2+ entry pathway (also known as capacitative Ca2+ entry; CCE). Fluxes across both plasma and ER membranes are in equilibrium. 2) Steady-state stimulated situation. After the initial release of Ca2+ from the ER, both Ca2+ pumps (SERCA and PMCA) are now fully activated and the store-operated Ca2+ entry path is also open, due to the reduced [Ca2+]ER. 3) Immediately after cessation of ACh stimulation. The IP3 receptor channels have closed and the SERCA pump can now refill the ER via Ca2+ entering through the plasma membrane. The PMCA is no longer activated because [Ca2+]i has quickly returned to the basal level. 4) Normal Ca2+ gradients have been restored to the resting basal level. [Adapted from (90).]
Local and Global Cytosolic Ca2+ Signals
In 1990, Osipchuk et al. (91) showed that a relatively high concentration of ACh causes synchronous oscillations of Ca2+-activated Cl− currents and [Ca2+]i, indicating that every time [Ca2+]i rises, the Cl− channels open. At low ACh concentration, however, there are repetitive short-lasting spikes of Ca2+-activated Cl− current, but no clear rise in the overall [Ca2+]i. Because the Ca2+-activated Cl− current spikes elicited by low-intensity ACh stimulation could be blocked by intracellular EGTA, it was concluded that these current spikes are due to repetitive cytosolic Ca2+ spikes in a small part of the cytosol close to the location of the Cl− channels. Because the spikes are local, it is understandable that they would not make a significant impact on the overall cytosolic Ca2+ concentration. Later that same year, Kasai and Augustine (92), using supramaximal ACh stimulation, demonstrated by Ca2+ imaging that the cytosolic Ca2+ signal always starts in the apical pole and then spreads as a wave towards the base of the cell. The initial apical Ca2+ signal is associated with Cl− current activation. Combining the information from these two papers of 1990, it appeared likely that the local Ca2+ spikes, activating the Cl− current spikes, are in the apical (secretory) pole.
In 1993, the Ca2+ spiking site was identified independently by two groups (93, 94). In these combined patch clamp and imaging experiments, it was shown that the repetitive Cl− current spikes elicited by low-intensity ACh stimulation were associated with cytosolic Ca2+ spikes confined exclusively to the apical (granular) pole. Furthermore, it was shown that flooding the cytosol with IP3 also elicited local Ca2+ spiking in the apical pole (93). Comparing the effects of locally injecting IP3 in the apical and basal parts of the cell, it was shown that the sensitivity of the Ca2+ release process to IP3 was far greater in the apical secretory pole than in the basal part of the cell (94). Almost ten years later, using a local uncaging approach to map the sensitivity of Ca2+-induced Ca2+ release (CICR), it was shown that CICR could be elicited in the apical pole, but not in the basal part of the cell (95).
Although studies carried out in the early 1990s imply that the Cl− channels are located in the apical membrane, this was only shown conclusively much later in another uncaging study, in which it was demonstrated that local uncaging of caged Ca2+ in the apical pole, but not in the lateral or basal parts of the cell, activates the Cl− current (96). Because Cl− channel activation is a critical step in transepithelial fluid secretion (84), the local apical Ca2+ spikes are physiologically important for this process. It was also shown, by capacitance measurements, that local apical Ca2+ signals are sufficient for triggering exocytosis (97). Thus, local apical Ca2+ signals trigger both fluid and enzyme secretion.
If IP3releases Ca2+from the ER Why Do the Ca2+ Signals Occur in the Granular Region?
The Ca2+ imaging studies in the early 1990s had established that the physiologically important cytosolic Ca2+ signals occur in the apical pole. All subsequent studies, including experiments in which the actions of the novel Ca2+ releasing messengers cyclic ADP-ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate (NAADP) were tested (98), have confirmed this. Nevertheless, many morphological studies had clearly shown that the ER is essentially confined to the basolateral part of the cell surrounding the nucleus, whereas the apical part is dominated by secretory (zymogen) granules (99). The generally accepted view that IP3 releases Ca2+ from the ER (100) therefore seemed to be in disagreement with the data from the pancreatic acinar cells. This problem has been difficult to solve, but the following experimental observations taken together provide an explanation: 1) Although most of the ER is located in the basolateral part, confocal studies of the distribution of fluorescent ER probes in normal living acinar cells show invasion of thin ER elements into the apical granular area (101). 2) Photobleaching/recovery studies show that the whole of the ER, including the ER elements in the granular part, is one continuous space and that Ca2+ can diffuse rapidly in the ER lumen (102). 3) The Ca2+ binding capacity of the cytosol is high (about 1500–2000), whereas the ER has a relatively low binding capacity (about 20). This is compatible with a model in which Ca2+ diffuses more easily in the ER than in the cytosol (103). 4) When the intracellular Ca2+ store has been emptied by supramaximal ACh stimulation in an externally Ca2+-free solution, the store can be refilled—in a thapsigargin-sensitive manner—from a point source (cell-attached patch pipette) at the base. This refilling occurs without any apparent rise in [Ca2+]i. Subsequent ACh stimulation elicits a normal cytosolic Ca2+ signal, initiated in the apical granular pole (104). 5) IP3 and cADPR, but not thapsigargin, release Ca2+ from isolated secretory (zymogen) granules (105). 6) Very recent studies on a novel two-photon permeabilized cell preparation, with well preserved function and polarity (Gerasimenko & Gerasimenko, in preparation), demonstrate that in the basal part of the cell there is only one type of Ca2+ store (ER), which includes the store in the nuclear envelope (106), whereas in the apical pole there is, in addition to the ER store, an acid thapsigargin- insensitive Ca2+ store, which is most likely in the secretory granules. All messengers tested (e.g., IP3, cADPR, and NAADP) can release Ca2+ from both the ER and the acid store. The Ca2+ tunnel model, in which Ca2+ taken up at the base is transferred across the cell through the ER lumen (Figure 9⇓), was originally proposed in 1997 on the basis of indirect studies, but is now supported by the considerable amount of experimental evidence described here.
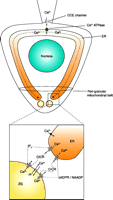
Ca2+signal generation in the apical granular pole. The main (upper) part shows a schematic diagram of a pancreatic acinar cell with the base at the top and the apical pole at the bottom. Ca2+ entry is depicted from a point source (cell-attached patch pipette) at the base. Ca2+ entering (through capacitative Ca2+ entry channels; CCE) is taken up by the ER Ca2+ pump and diffuses in the lumen of the ER towards the apex. At the apical pole, Ca2+ can be released through IP3 receptors and ryanodine channels, which can be stimulated, respectively, by IP3 and cADPR or NAADP (acting via different intermediary receptor proteins, but both ultimately activating the ryanodine receptor). Ca2+ released from the ER terminals can induce further Ca2+ release (CICR) from the secretory (zymogen granules; ZG), assisted by IP3 and cADPR/NAADP. The cytosolic Ca2+ signals are confined to the apical pole by the peri-granular mitochondrial belt. [Adapted and up-dated from (104).]
The Mitochondrial Firewall
The high cytosolic Ca2+ buffer capacity in acinar cells, almost certainly due to bound or very high molecular–weight buffers, will help to reduce dispersion of Ca2+ signals generated in the apical pole. Even so, it may be difficult to understand how, in such a relatively small cell (diameter ~20μm), it is possible to have relatively long-lasting (10–20 seconds) standing Ca2+ gradients between the apical and basal parts of the cell. This mystery was solved in 1999, when it was shown that there is a major concentration of mitochondria in a belt surrounding the apical granular region, separating it from the basolateral part of the cell (107); the concentration of mitochondria is particularly high close to the apical membrane. The mitochondrial belt serves as a Ca2+ buffer barrier, taking up Ca2+ rapidly during an increase in the apical cytosolic Ca2+ concentration (local spike) and releasing the Ca2+ taken up much more slowly (108). It has been shown that inhibition of mitochondrial function transforms IP3- elicited repetitive local Ca2+ spikes to a global and sustained Ca2+ elevation (107).
The perigranular mitochondrial belt does not only serve as a Ca2+ buffer barrier; a rise in the mitochondrial Ca2+ concentration also increases metabolism via Ca2+-activated enzymes in the Krebs cycle. Recordings of [Ca2+] in the cytosol and the mitochondria together with measurement of the mitochondrial NADH concentration have shown that a rise in the cytosolic Ca2+ concentration is followed quickly by a rise in the mitochondrial Ca2+ concentration, which in turn is followed by a rise in the NADH concentration (109). There are two other physiologically important mitochondrial localizations: around the nucleus and just below the surface cell membrane (108). In general, the three groups of mitochondria described here divide the cytosol into sub-compartments in which the cytosolic Ca2+ concentration can be regulated separately, there-by allowing specific localized control of this important messenger.
Physiological and Pathological Release of Ca2+from the ER
Physiological stimulation of the acinar cells causes mainly repetitive local Ca2+ spikes in the apical granular pole and only occasionally more prolonged (global) transients. The lower the agonist concentration, the lower is the probability of the short-lasting local spikes triggering a more prolonged global transient. Of the two physiological agonists ACh and cholecystokinin (CCK), CCK is more likely to trigger global Ca2+ transients, but at most physiological CCK concentrations (2–5 pM), the short-lasting apical spikes are by far the most frequent (98). The short-lasting local cytosolic Ca2+ spikes are not associated with any measurable reduction in the overall [Ca2+]ER, whereas a prolonged and global cytosolic Ca2+ transient is due to a substantial liberation of Ca2+ from the ER, as seen by a clear transient drop in [Ca2+]ER (102). Prolonged supramaximal stimulation causes complete emptying of Ca2+ from the ER (102). Such stimulation is associated with activation of trypsinogen in the granular region and transformation of the normally electron-dense granules into empty-looking vacuoles. Both these processes are Ca2+-dependent (110) and are similar to what happens in human acute pancreatitis (i.e., the pancreas digests itself). The most common causes of pancreatitis are alcohol abuse and gall bladder stones with reflux of bile into the pancreas. In this context, it is interesting that bile acids in pathophysiologically relevant concentrations cause prolonged global cytosolic Ca2+ signals and Ca2+-dependent necrosis (111, 112) and that several fatty acid ethyl esters and fatty acids, important non-oxidative alcohol metabolites, also cause sustained global [Ca2+]i elevations due to complete emptying of the ER. This is also associated with Ca2+-dependent necrosis (113). Taken together, these data indicate that normal secretion control is due to local apical Ca2+ spikes, which can be generated with minimal reductions in [Ca2+]ER, whereas complete emptying of the ER Ca2+ store, causing a sustained [Ca2+]i elevation, is associated with necrosis.
Acknowledgments
This article stems from an Experimental Biology 2005 symposium sponsored by the Division of Systems and Integrative Pharmacology of ASPET. The symposium celebrates the seminal work of Roger Tsien and colleagues (1), cited nearly 15,500 times, advancing the use of fluorescent dyes to measure intracellular ions and other physiological parameters. Support for WFG is from the Austrian Science Funds (P-16860-B9 and SFB714) and the infrastructure programs (UGP4) of the Austrian ministry of education, science and culture.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Ole H. Petersen, FRS, is Medical Research Council Professor in Physiology, at The University of Liverpool, UK.

Wolfgang F. Graier, PhD, is Associate Professor of Physiology and Biochemical Pharmacology at the Institute of Molecular BIology and Biochemistry of the Center for Molecular Medicine, Medical University of Graz, Austria.

W. Jonathan Lederer, MD, PhD, is Professor of Physiology and Director of the Medical Biotechnology Center at the University of Maryland School of Medicine in Baltimore.

Ismail Laher, PhD, and Harm J. Knot (not shown) are the organizers of the Experimental Biology 2005 symposium on which this article is based. Ismail Laher is Associate Professor of Pharmacology and Therapeutics at the University of British Columbia. Harm J. Knot is Assistant Professor of Pharmacology and Therapeutics at the University of Florida College of Medicine in Gainesville. Address correspondence to IL. E-mail ilaher{at}interchange.ubc.ca.

