|
Show all abstracts Show selected abstracts Add to my list |
|
| EDITORIAL |
|
|
|
Methylenetetrahydrofolatereductase C677T polymorphism and folate metabolism in human health |
p. 99 |
Babu Rao Vundinti
DOI:10.4103/0971-6866.142840 PMID:25400337 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| REVIEW ARTICLES |
 |
|
|
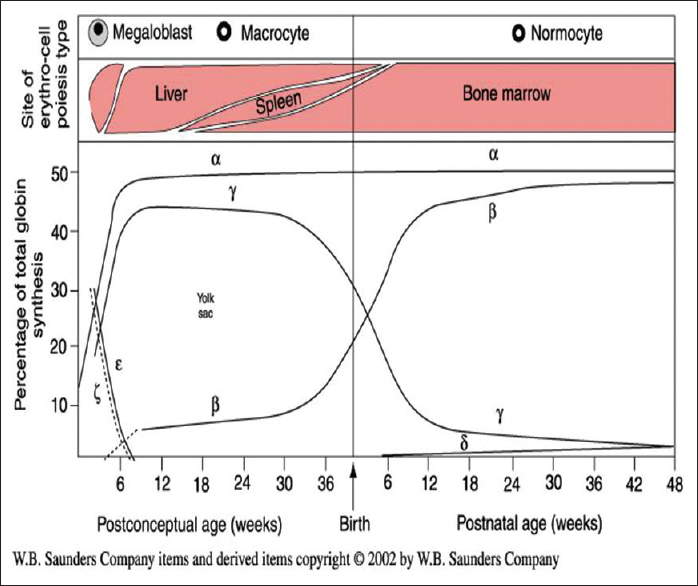  |
Guidelines for screening, diagnosis and management of hemoglobinopathies  |
p. 101 |
Kanjaksha Ghosh, Roshan Colah, Mamta Manglani, Ved Prakash Choudhry, Ishwar Verma, Nishi Madan, Renu Saxena, Dipty Jain, Neelam Marwaha, Reena Das, Dipika Mohanty, Rajendra Choudhary, Sarita Agarwal, Malay Ghosh, Cecil Ross
DOI:10.4103/0971-6866.142841 PMID:25400338The β-thalassemias and sickle cell disorders are a major health burden in India. Diagnosis and management of these disorders both in adults and in newborns using appropriate approaches and uniform technology are important in different regions of a vast and diverse country as India. In view of a National Thalassemia Control Program to be launched soon, a need was felt for guidelines on whom to screen, cost-effective technologies that are to be used as well as for establishing prenatal diagnosis programs in regional centers. Newborn screening for sickle cell disorders is in its infancy in India and uniform approaches need to be followed. Also, included are guidelines for monitoring and managing patients who are now growing older and need comprehensive care as well as management of complications of the disease. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Genetics in psychiatry |
p. 120 |
Shreekantiah Umesh, Shamshul Haque Nizamie
DOI:10.4103/0971-6866.142845 PMID:25400339Today, psychiatrists are focusing on genetics aspects of various psychiatric disorders not only for a future classification of psychiatric disorders but also a notion that genetics would aid in the development of new medications to treat these disabling illnesses. This review therefore emphasizes on the basics of genetics in psychiatry as well as focuses on the emerging picture of genetics in psychiatry and their future implications. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
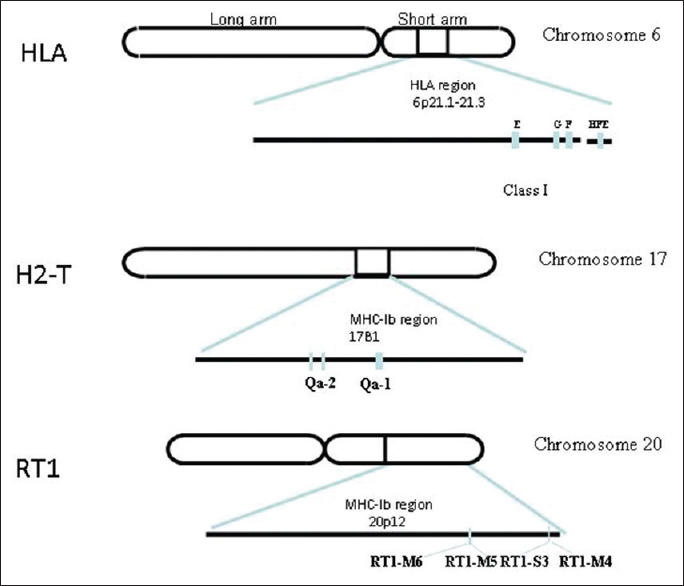 |
Mammalian non-classical major histocompatibility complex I and its receptors: Important contexts of gene, evolution, and immunity |
p. 129 |
BM Pratheek, Tapas K Nayak, Subhransu S Sahoo, Prafulla K Mohanty, Soma Chattopadhyay, Ntiya G Chakraborty, Subhasis Chattopadhyay
DOI:10.4103/0971-6866.142855 PMID:25400340The evolutionary conserved, less-polymorphic, nonclassical major histocompatibility complex (MHC) class I molecules: Qa-1 and its human homologue human leukocyte antigen-E (HLA-E) along with HLA-F, G and H cross-talk with the T-cell receptors and also interact with natural killer T-cells and other lymphocytes. Moreover, these nonclassical MHC molecules are known to interact with CD94/NKG2 heterodimeric receptors to induce immune responses and immune regulations. This dual role of Qa-1/HLA-E in terms of innate and adaptive immunity makes them more interesting. This review highlights the new updates of the mammalian nonclassical MHC-I molecules in terms of their gene organization, evolutionary perspective and their role in immunity. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| ORIGINAL ARTICLES |
|
|
|
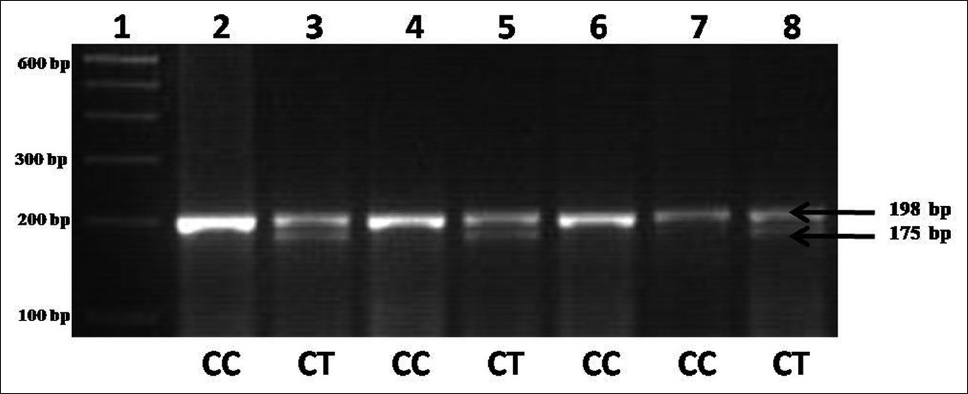 |
Methylenetetrahydrofolate reductase polymorphism is not risk factor for Down syndrome in North India |
p. 142 |
Vandana Rai, Upendra Yadav, Pradeep Kumar, Sushil Kumar Yadav
DOI:10.4103/0971-6866.142858 PMID:25400341Background: Down syndrome (DS) is the most common cause of mental retardation of genetic etiology with the prevalence rate of 1/700 to 1/1000 live births worldwide. Several polymorphisms in folate/homocysteine metabolism pathways genes have been reported as a risk factor in women for bearing DS child, but very few studies investigated these polymorphisms in DS cases whether there are a risk factor for being DS or not.
Objective: We have investigated the association of methylenetetrahydrofolate reductase (MTHFR) with the occurrence of DS in Indian population. MTHFR is one of the key regulatory enzymes involved in the metabolic pathway of homocysteine responsible for the reduction of methyltetrahydrofolate. A total of 32 DS cases and 64 age, sex matched controls were genotyped for MTHFR C677T polymorphism by polymerase chain reaction-restriction fragment length polymorphism.
Results: The observed genotype frequencies were CC = 0.81; CT = 0.17 and TT = 0.02 in controls and CC = 0.81 and CT = 0.19 in DS cases. Frequency of T allele in DS and controls were 0.09 and 0.1, respectively. Significant difference in the distribution of mutant 677T allele was not observed between DS cases and controls (odds ratio = 0.915; 95% confidence intervals: 0.331-2.53; P = 0.864).
Conclusion: Results of this study indicate that MTHFR C677T polymorphism is not risk factor for DS. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
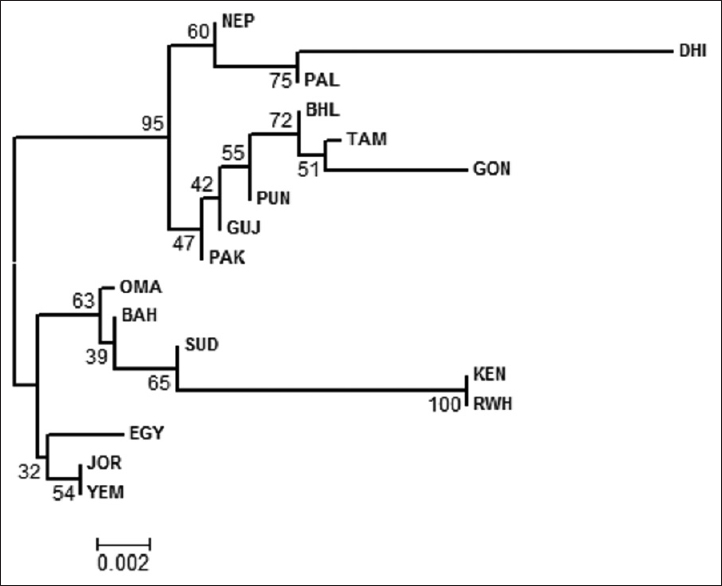 |
Genetic diversity of 15 autosomal short tandem repeats loci using the AmpFLSTR ® Identifiler™ kit in a Bhil Tribe Population from Gujarat state, India |
p. 148 |
Ramesh R Chaudhari, MS Dahiya
DOI:10.4103/0971-6866.142879 PMID:25400342Materials and Methods: The genetic diversity and forensic parameters based on 15 autosomal short tandem repeats (STR) loci; D8S1179,D21S11, D7S820, CSF1PO, D3S1358, TH01, D13S317,D16S539, D2S1338, D19S433, vWA, TPOX, D18S51,D5S818, and FGA in AmpFLSTR® Identifiler™ kit from Applied Biosystems, Foster City, CA, USA were evaluated in saliva samples of 297 unrelated individuals from the Bhil Tribe population of Gujarat state, India to study genetic diversities and relatedness of this population with other national and international populations.
Resuits: Statistical analysis of the data revealed all loci were within Hardy-Weinberg Equilibrium expectations with the exception of the locus vWA (0.019) and locus D18S51 (0.016). The neighbour joining phylogeny tree and Principal Co-ordinate Analysis plot constructed based on Fst distances from autosomal STRs allele frequencies of the present study and other national as well as international populations show clustering of all the South Asian populations in one branch of the tree, while Middle Eastern and African populations cluster in a separate branch.
Conclusion: Our findings reveal strong genetic affinities seen between the Indo-European (IE) speaking Bhil Tribe of Gujarat and Dravidian groups of South India. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Cost-effectiveness analysis for triple markers serum screening for Down's syndrome in Thai setting |
p. 153 |
Viroj Wiwanitkit
DOI:10.4103/0971-6866.142880 PMID:25400343Background: Down's syndrome is an important congenital chromosomal disorder that can be seen around the world. The antenatal screening for this disorder is an important processing in present obstetrics.
Objective: Due to the concept of first do no harm, the use of noninvasive test is recommended. The triple marker screening test has been introduced for a few years and acceptable for its effi cacy.
Result: However, an important concern is on its cost-effectiveness. Here, the author analyze and present the cost-effectiveness of the triple markers serum screening for Down's syndrome in Thai setting.
Conclusion: According to this work, the cost per effectiveness of triple markers serum screening is slightly lower than standard amniocentesis test. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Methylenetetrahydrofolate reductase C677T variant in Indian children with craniosynostosis: Its role in the pathogenesis, risk of craniosynostosis |
p. 155 |
Rajeev Kumar Pandey, Abid Ali, Amit Singh, Sukanya Gayan, Minu Bajpai
DOI:10.4103/0971-6866.142882 PMID:25400344Background: 677C to T allele in the 5, 10-methylenetetrahydrofolate reductase (MTHFR) gene has been implicated in the etiology of various syndromes and nonsyndromic diseases but till date no direct studies have been reported with craniosynostosis.
Objectives: The aim was to study the family-based association of MTHFR polymorphism in different categories of craniosynostosis patients.
Materials and Methods: This was a cross-sectional study in which 30 patients classified as Apert syndrome, Pfeiffr syndrome and nonsyndromic craniosynostosis patients with their family were recruited. A sample of 3 ml intravenous blood was taken from patients and from their family members (father and mother) in ethylenediaminetetraacetic acid-anticoagulated vacutainer for the purpose of the study. Genomic DNA was extracted from peripheral blood lymphocytes by phenol chloroform extraction method. Primers for MTHFR gene were designed. The polymerase chain reaction was carried out. After successful amplification, a small aliquot (5 μl) of the MTHFR reaction mixture was treated with 1 units of Hinf I restriction enzyme (NEB). Results were obtained and compiled.
Results: A total of 30 patients/participants with craniosynostosis of Indian descent and their parents formed the study group. The genotyping did not confirm an association between the MTHFR 677C to T polymorphism and between different categories of craniosynostosis. When comparing the offspring of mothers statistically significant differences were found.
Conclusion: C667T polymorphism of the MTHFR gene is unlikely to play a role in the pathogenesis of craniosynostosis though maternal MTHFR C677T polymorphism may be a genetic risk factor. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
FLT3 and NPM-1 mutations in a cohort of acute promyelocytic leukemia patients from India |
p. 160 |
Suchitra Swaminathan, Swati Garg, Manisha Madkaikar, Maya Gupta, Farah Jijina, Kanjaksha Ghosh
DOI:10.4103/0971-6866.142884 PMID:25400345Background: Acute promyelocytic leukemia (APL) with t (15;17) is a distinct category of acute myeloid leukemia (AML) and is reported to show better response to anthracyclin based chemotherapy. A favorable overall prognosis over other subtypes of AML has been reported for APL patients but still about 15% patients relapse.
Methods: This study evaluated the presence of Famus like tyrosine kinase-3 (FLT3) and nucleophosmin-1 (NPM1) gene mutations in a cohort of 40 APL patients. Bone marrow/peripheral blood samples from patients at the time of diagnosis and follow-up were processed for immunophenotyping, cytogenetic markers and isolation of DNA and RNA. Samples were screened for the presence of mutations in FLT3 and NPM1 genes using polymerase chain reaction followed by sequencing.
Results: Frequency of FLT3/internal tandem duplication and FLT3/tyrosine kinase domain was found to be 25% and 7% respectively. We observed a high frequency of NPM1 mutation (45%) in the present population of APL patients. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
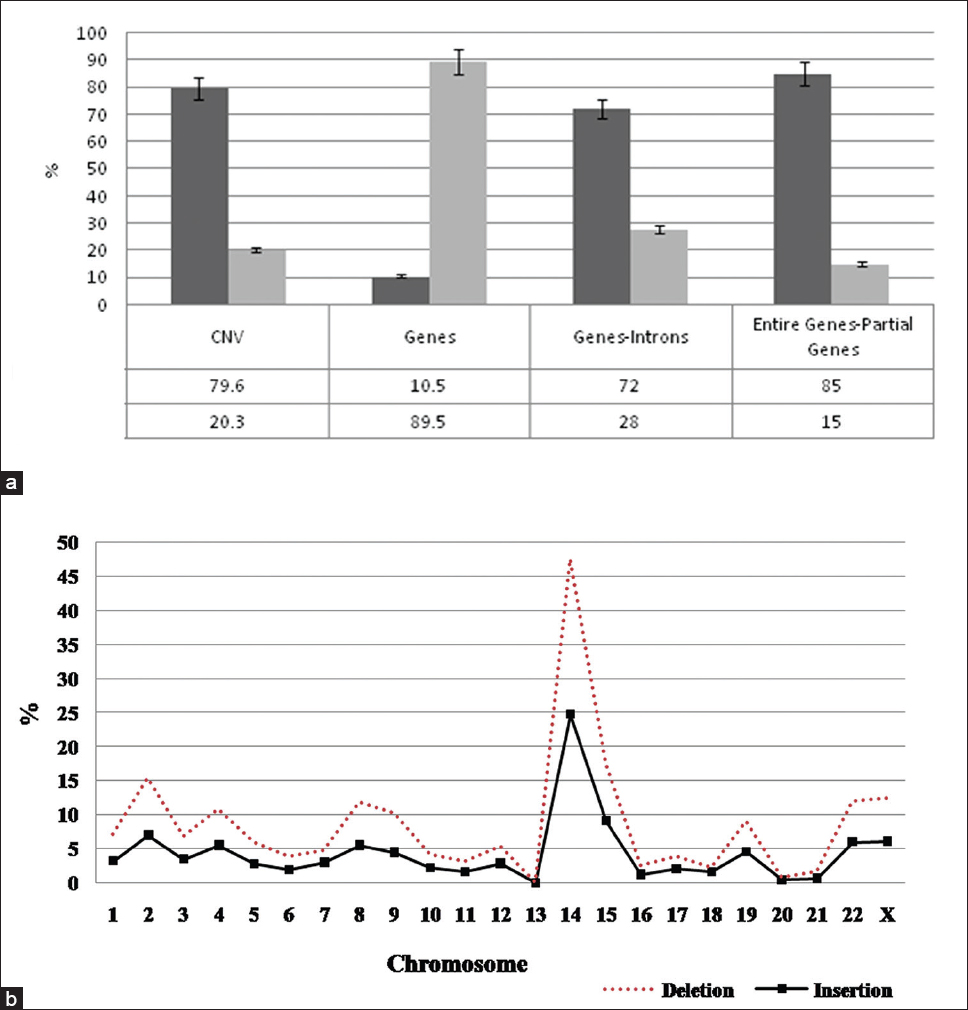 |
Insertion-deletions burden in copy number polymorphisms of the Tibetan population |
p. 166 |
Avinash M Veerappa, Sangeetha Vishweswaraiah, Kusuma Lingaiah, N Megha Murthy, Raviraj V Suresh, Keshava Belur, Nallur B Ramachandra, Tejaswini , Niveditha B Patel, PK Supriya Gowda
DOI:10.4103/0971-6866.142888 PMID:25400346Background: Many studies have been conducted to identify either insertions-deletions (inDels) or copy number variations (CNVs) in humans, but few studies have been conducted to identify both of these forms coexisting in the same region.
Aims and Objectives: To map the functionally significant sites within human genes that are likely to influence human traits and diseases.
Materials and Methods: In this report, we describe an inDel map in the 1051 Tibetan CNV regions obtained through CNV genotyping using Affymetrix Genome-wide single nucleotide polymorphism 6.0 chip. InDel polymorphisms in these copy number polymorphism regions were identified with a computational approach using the 2500 deoxyribonucleic acid sequences obtained from the 1000 Genome Project.
Results: The study identified a total of 95935 inDels that range from 1 bp to several bps in length which were found scattered across regulatory regions, exons and in introns of genes underlying the CNVs. A study on the distribution of inDels revealed that the majority of inDels were found in coding regions of the genome than the noncoding, while within the genes, inDels in intron regions were more followed by exonic regions and finally the regulatory regions.
Conclusion: Study of inDels in CNV regions contribute to the enhanced understanding of the role played by the two variations and their collective influence on the genome. Further, a collection of these inDel genetic markers will aid in genetic mapping, further understanding of the phenotypic variability, identification of disease genes and in detecting novel CNVs. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
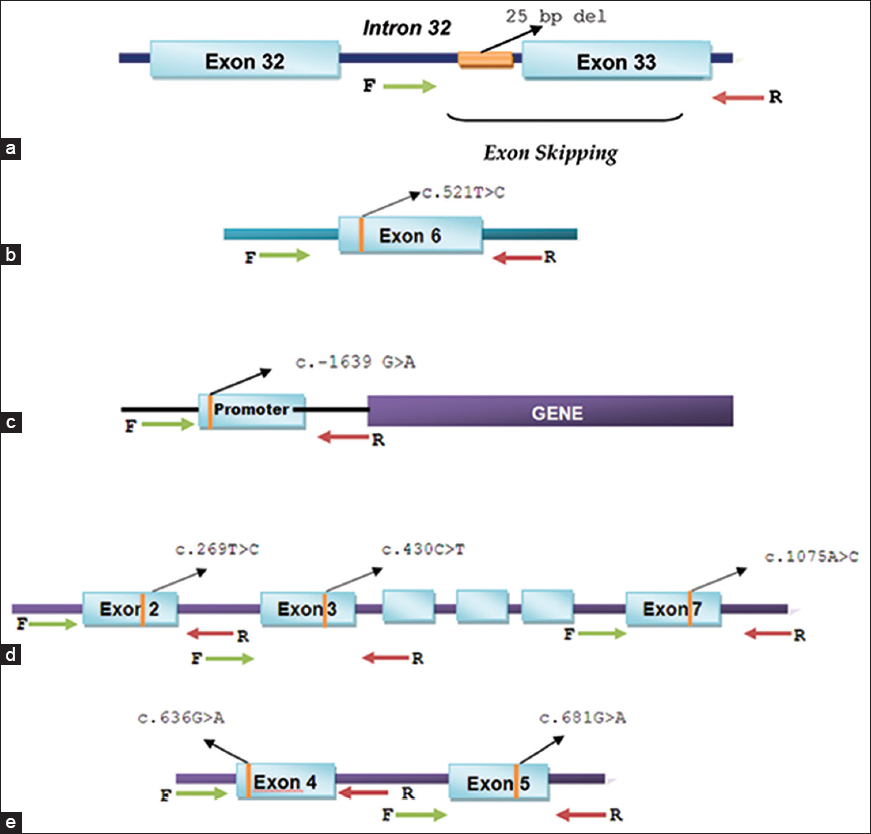 |
Prevalence of genetic variants associated with cardiovascular disease risk and drug response in the Southern Indian population of Kerala |
p. 175 |
Lakshmi Mahadevan, Ancy Yesudas, PK Sajesh, S Revu, Prasanna Kumar, Devi Santhosh, Sam Santhosh, JM Sashikumar, VK Gopalakrishnan, Joji Boben, Changanamkandath Rajesh
DOI:10.4103/0971-6866.142896 PMID:25400347Background and Aim: This study reports the prevalence of five clinically significant variants associated with increased risk of cardiovascular disorders, and variable responses of individuals to commonly prescribed cardiovascular drugs in a South Indian population from the state of Kerala.
Materials and Methods: Genomic DNA isolated from 100 out-patient samples from Kerala were sequenced to examine the frequency of clinically relevant polymorphisms in the genes MYBPC3 (cardiomyopathy), SLCO1B1 (statin-induced myopathy), CYP2C9, VKORC1 (response to warfarin) and CYP2C19 (response to clopidogrel).
Results: Our analyses revealed the frequency of a 25 bp deletion variant of MYBPC3 associated with risk of cardiomyopathy was 7%, and the SLCO1B1 "C" allele associated with risk for statin-induced myopathy was 15% in this sample group. Among the other variants associated with dose-induced toxicity of warfarin, VKORC1 (c.1639G>A), was detected at 22%, while CYP2C9*3 and CYP2C9*2 alleles were present at a frequency of 15% and 3% respectively. Significantly, the tested sample population showed high prevalence (66%) of CYP2C19*2 variant, which determines response to clopidogrel therapy.
Conclusions: We have identified that certain variants associated with cardiovascular disease and related drug response in the five genes, especially those in VKORC1, CYP2C19 and MYBPC3, are highly prevalent in the Kerala population, with almost 2 times higher prevalence of CYP2C19*2 variant compared with other regions in the country. Since the variants chosen in this study have relevance in disease phenotype and/or drug response, and are detected at a higher frequency, this study is likely to encourage clinicians to perform genetic testing before prescribing therapy. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| CASE REPORTS |
|
|
|
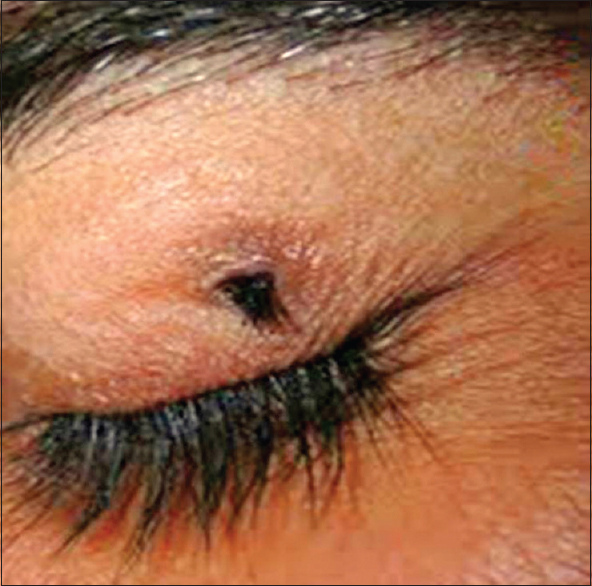 |
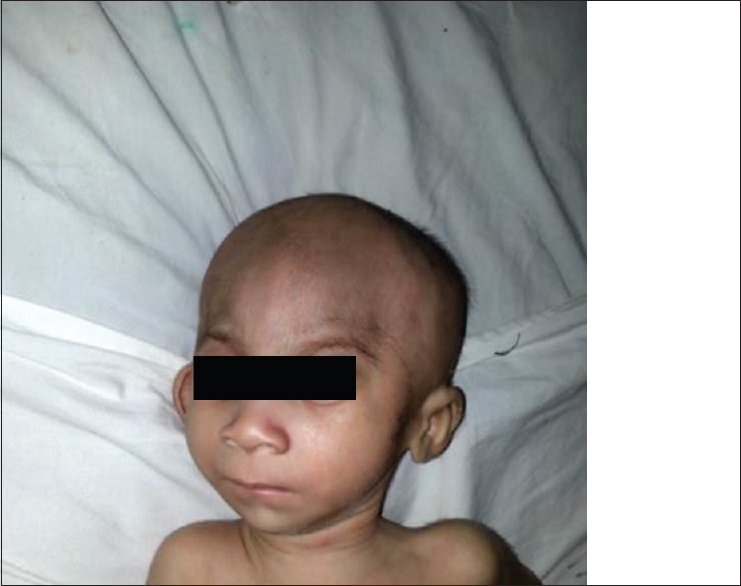
Ectopia cilia with pedigree analysis: Second case report in the world |
p. 185 |
Tarang Goyal, Anupam Varshney, SK Bakshi
DOI:10.4103/0971-6866.142897 PMID:25400348We present a case of ectopia cilia in a 28-year-old male patient. Ectopia cilia was were seen in the outer third of left upper eyelid. The patient's maternal grandfather also had ectopia cilia of the left upper eyelid as reported by the patient's mother. Ectopia cilia is a rare condition seen in humans. Only 12 cases of ectopic cilia in humans have been reported so far in the world. The present case of ectopia cilia is the second case report in the world with pedigree analysis. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Fetal valproate syndrome |
p. 187 |
Parmarth G Chandane, Ira Shah
DOI:10.4103/0971-6866.142898 PMID:25400349Antenatal use of anticonvulsant valproic acid can result in a well-recognized cluster of facial dysmorphism, congenital anomalies and neurodevelopmental retardation. In this report, we describe a case with typical features of fetal valproate syndrome (FVS). A 26-year-old female with epilepsy controlled on sodium valproate 800 mg/day since 3 years, gave birth to a male child with characteristic features of FVS. She also had 3 spontaneous first-trimester abortions during those 3 years. Sodium valproate, a widely used anticonvulsant and mood regulator, is a well-recognized teratogen that can result in facial dysmorphism, craniosynostosis, neural tube defects, and neurodevelopmental retardation. Therefore, we strongly recommend avoidance of valproic acid and supplementation of folic acid during pregnancy. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
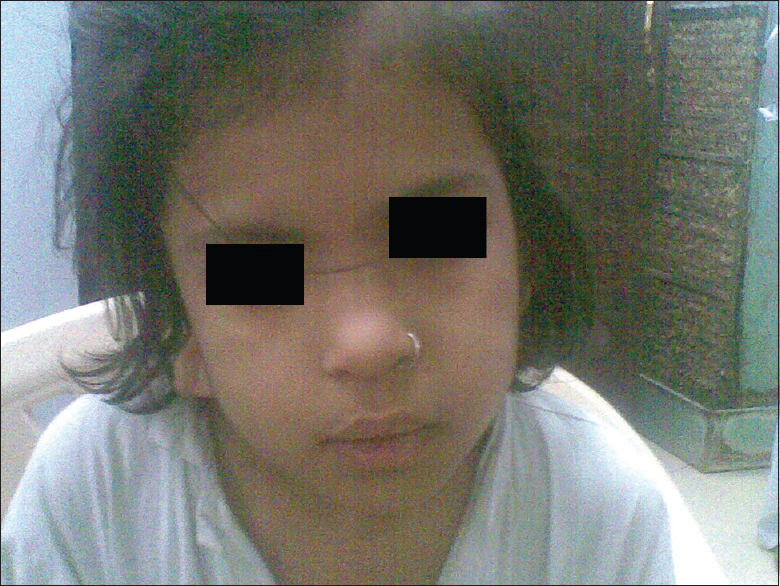 |
Wildervanck syndrome with hypoplastic frontal sinus: A rare case presentation |
p. 189 |
Suwansh Sukhadeorao Meshram, Sheetal Nikose, Shraddha Jain, Amar Taksande
DOI:10.4103/0971-6866.142899 PMID:25400350We report a case of Wildervanck syndrome exhibiting Klippel-Feil anomaly, Duane's retraction syndrome and congenital deafness. Since the first case was reported in 1952, there have been more reports describing this triad either complete or incomplete. Our case has a complete triad of the syndrome along with frontal sinus hypoplasia. Our case is unique as the triad was associated with frontal sinus hypoplasia, which is very rare association. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
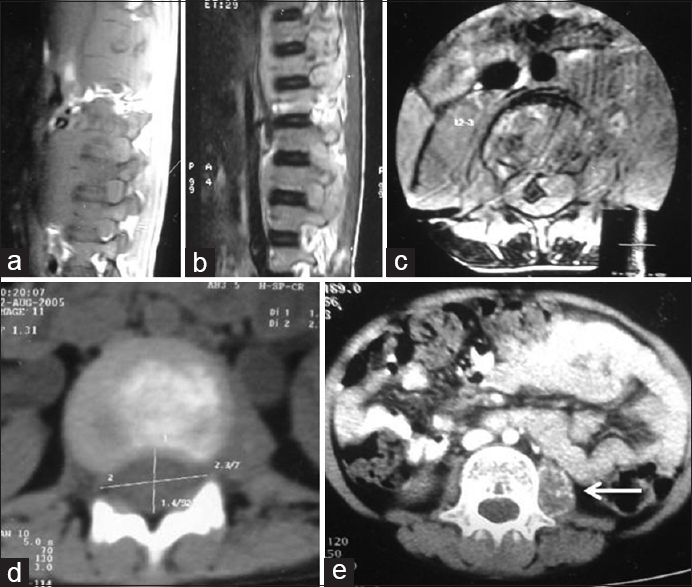 |
Constitutional mismatch repair deficiency syndrome: Do we know it? |
p. 192 |
C Ramachandra, Vasu Reddy Challa, Rachan Shetty
DOI:10.4103/0971-6866.142902 PMID:25400351Constitutional mismatch repair deficiency syndrome is a rare autosomal recessive syndrome caused by homozygous mutations in mismatch repair genes. This is characterized by the childhood onset of brain tumors, colorectal cancers, cutaneous manifestations of neurofibromatosis-1 like café au lait spots, hematological malignancies, and occasionally other rare malignancies. Here, we would like to present a family in which the sibling had glioblastoma, and the present case had acute lymphoblastic lymphoma and colorectal cancer. We would like to present this case because of its rarity and would add to literature. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
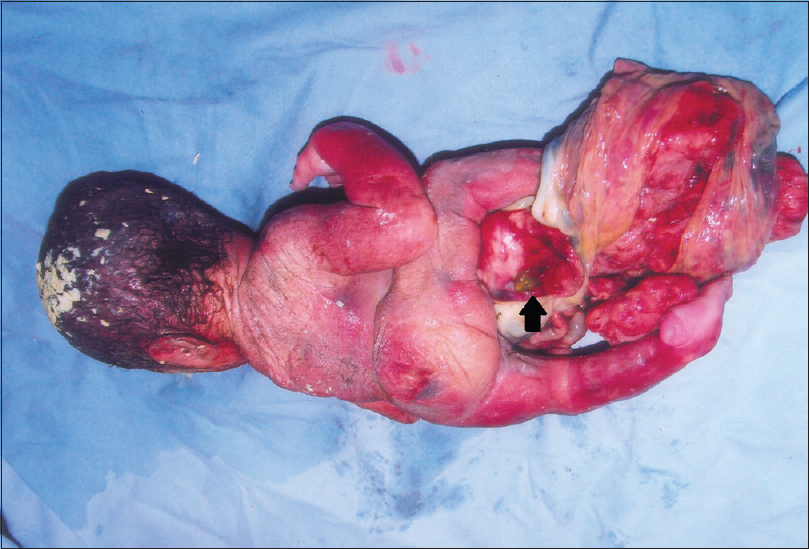 |
Omphalocele, exstrophy of cloaca, imperforate anus and spinal defect (OEIS Complex) with overlapping features of body stalk anomaly (limb body wall complex) |
p. 195 |
Suresh R. S. Mandrekar, Sangeeta Amoncar, Siddhartha Banaulikar, Vishal Sawant, R. G. W. Pinto
DOI:10.4103/0971-6866.142906 PMID:25400352OEIS is an extremely rare constellation of malformations, which includes omphalocele, exstrophy of cloaca, imperforate anus, and spinal defect. We report here autopsy findings in a case of OEIS complex, which apart from the major anomalies of the complex had bilateral club foot that is, congenital talipes equinovarus, right hydroureter, and body stalk anomaly. The umbilical cord was absent, and the umbilical vessels were embedded in an amniotic sheet, which connected the skin margin of the anterior body wall defect to the placenta, this feature being the hallmark of limb body wall complex (LBWC). This case further supports the view that OEIS and LBWC represent a continuous spectrum of abnormalities rather than separate conditions and may share a common etiology and pathogenetic mechanism as proposed by some authors. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
First report of c. 1499G>C mutation in a 6-month-child with cystic fibrosis |
p. 199 |
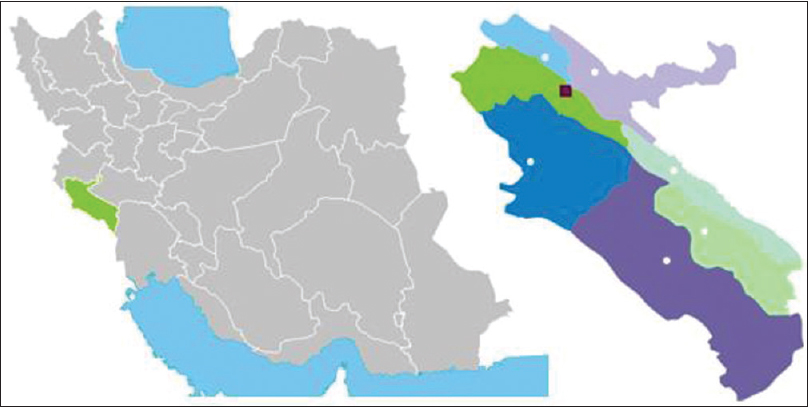
Abbas Sahami, Nourkhoda Sadeghifard, Alireza Monsef, Hadi Peyman
DOI:10.4103/0971-6866.142911 PMID:25400353So far, more than 1800 mutations identified in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. In this case report, we presented first report of c. 1499G>C mutation in a 6-month-old girl with cystic fibrosis (CF) diagnosis. A 6-month-old girl with weakness and meconium Ileus referred to the pediatric clinic in Ilam, in the west of Iran. Patient's skin was dark and suffered from bronchiectasis. The sweat test was performed, and the concentration of chloride and sodium in patient's sweat was 130-135 mmol/L and 125-128 mmol/L, respectively. The exon 10 mutation analysis of a CF patient was performed. CFTR mutation analysis revealed the identification of 2 mutations in patient, the mutations were p.F508del (ΔF508) and c. 1499G>C (cd500), respectively. The mutation c. 1499G>C (cd500) were found for the first time in the world. Assessing this mutation in future study and genetic investigation is recommended. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Phenotypical characterization of 13q deletion syndrome: Report of two cases |
p. 203 |
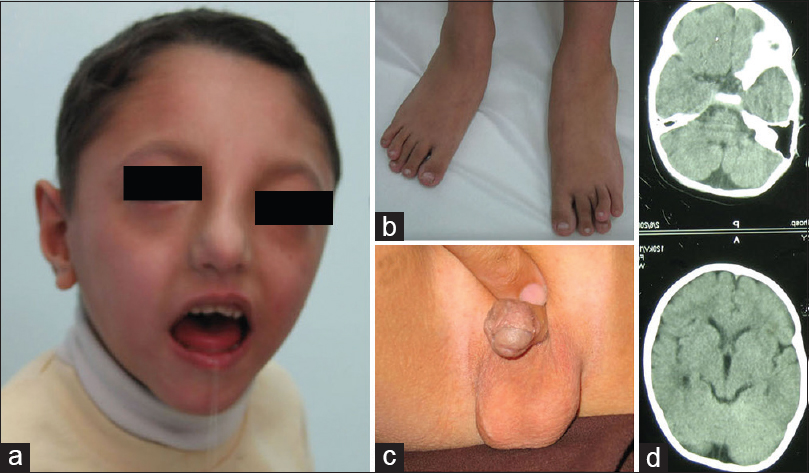
Eiman Bagherizadeh, Yousef Shafaghati, Fatemeh Hadipour, Farkhondeh Behjati
DOI:10.4103/0971-6866.142912 PMID:25400354Patients with 13q deletion syndrome are characterized with different phenotypical features depending on the size and location of the deleted region on chromosome 13. These patients fall into three groups: In Group 1, deleted region is in the proximal and does not extend into q32; in Group 2, deleted region involves proximal to the q32 and in Group 3 q33-q34 is deleted. We present two cases with 13q syndrome with two different deleted region and different severity on clinical features: One case with interstitial deletion belongs to the Group 1 with mild mental retardation and minor malformations and the other case with terminal deletion belongs to Group 3 with moderate to severe mental retardation and major malformations. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Congenital anonychia and brachydactyly of the left foot - Cooks syndrome variant: Case report and review of literature |
p. 206 |
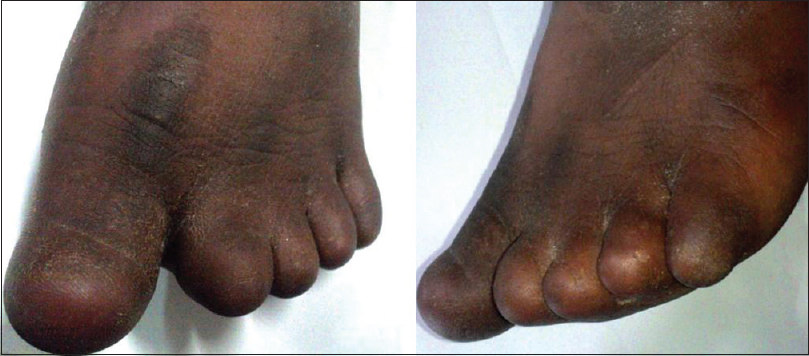
Daipayan Chatterjee
DOI:10.4103/0971-6866.142914 PMID:25400355Cooks syndrome is characterized by familial congenital anonychia or onychodystrophy, hypoplasia or absence of distal phalanges of the hands and feet with brachydactyly of the fifth finger and digitalization of the thumb (triphalangism). It is listed as a "rare disease" by the Office of Rare Diseases of the National Institutes of Health. Here, we report a case of congenital anonychia and brachydactyly of the left foot, which possibly is a variant of Cooks syndrome with a positive family history of similar deformity. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| LETTER TO THE EDITOR |
|
|
|
|
Association of maternal folate with methylene tetrahydrofolate reductase polymorphism relationship in infants <3 months with Down syndrome |
p. 209 |
Pankaj Kumar Mohanty, Seema Kapoor
DOI:10.4103/0971-6866.142915 PMID:25400356 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|










