|
Show all abstracts Show selected abstracts Add to my list |
|
| EDITORIAL |
|
|
|
Explaining anthropometric variations in sickle cell disease requires a multidimensional approach |
p. 1 |
Malay B Mukherjee, Kanjaksha Ghosh
DOI:10.4103/0971-6866.96632 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| COMMENTARY |
 |
|
|
  |
Recollections of J.B.S. Haldane, with special reference to Human Genetics in India |
p. 3 |
Krishna R Dronamraju
DOI:10.4103/0971-6866.96634 This paper is a brief account of the scientific work of J.B.S. Haldane (1892-1964), with special reference to early research in Human Genetics. Brief descriptions of Haldane's background, his important contributions to the foundations of human genetics, his move to India from Great Britain and the research carried out in Human Genetics in India under his direction are outlined. Population genetic research on Y-linkage in man, inbreeding, color blindness and other aspects are described. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| REVIEW ARTICLES |
|
|
|
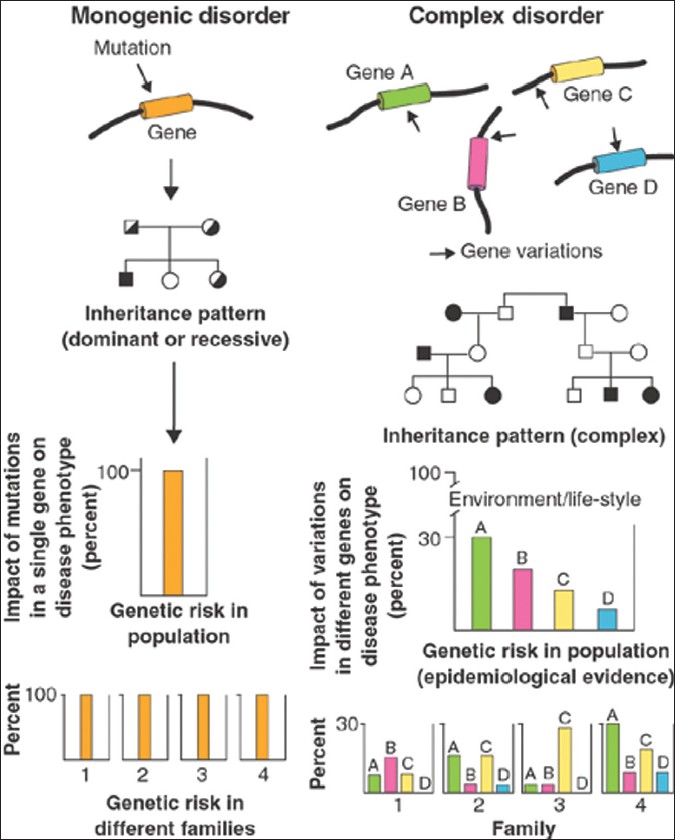 |
Effect of gene polymorphisms on periodontal diseases |
p. 9 |
Fouzia Tarannum, Mohamed Faizuddin
DOI:10.4103/0971-6866.96638 Periodontal diseases are inflammatory diseases of supporting structures of the tooth. It results in the destruction of the supporting structures and most of the destructive processes involved are host derived. The processes leading to destruction and regeneration of the destroyed tissues are of great interest to both researchers and clinicians. The selective susceptibility of subjects for periodontitis has remained an enigma and wide varieties of risk factors have been implicated for the manifestation and progression of periodontitis. Genetic factors have been a new addition to the list of risk factors for periodontal diseases. With the availability of human genome sequence and the knowledge of the complement of the genes, it should be possible to identify the metabolic pathways involved in periodontal destruction and regeneration. Most forms of periodontitis represent a life-long account of interactions between the genome, behaviour, and environment. The current practical utility of genetic knowledge in periodontitis is limited. The information contained within the human genome can potentially lead to a better understanding of the control mechanisms modulating the production of inflammatory mediators as well as provides potential therapeutic targets for periodontal disease. Allelic variants at multiple gene loci probably influence periodontitis susceptibility. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (6) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Genetic biomarkers of depression |
p. 20 |
Anand Tamatam, Farhath Khanum, Amarinder Singh Bawa
DOI:10.4103/0971-6866.96639 Depression is a term that has been used to describe a variety of ailments, ranging from minor to incapacitating. Clinically significant depression, termed as major depression, is a serious condition characterized not only by depressed mood but also by a cluster of somatic, cognitive, and motivational symptoms. Significant research efforts are aimed to understand the neurobiological as well as psychiatric disorders, and the evaluation of treatment of these disorders is still based solely on the assessment of symptoms. In order to identify the biological markers for depression, we have focused on gathering information on different factors responsible for depression including stress, genetic variations, neurotransmitters, and cytokines and chemokines previously suggested to be involved in the pathophysiology of depression. The present review illustrates the potential of biomarker profiling for psychiatric disorders, when conducted in large collections. The review highlighted the biomarker signatures for depression, warranting further investigation. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Science of breeding and heredity from ancient Persia to modern Iran |
p. 34 |
Mohammad H Kariminejad, Ardeshir Khorshidian
DOI:10.4103/0971-6866.96641 About 1700 years BC, the prophet Zoroaster declared equal right for women and men to choose their "own ways." There is much evidence that ancient Persians believed in the equal contribution of women and men toward producing a child, and all its hereditary characteristics.
Even more surprising are the phrases in Vandidad book, which were gathered by Mobedans in the Mad dynasty about egg extraction (gametes) from animal reproductive organs (gonads) and their storage for future conception.
Centuries later, Western philosopher beliefs in regard to reproduction were contrary to Persian knowledge. The Greek philosophers believed that man's water (semen) contains all human characteristics, and the female uterus is only responsible for nurturing and development of fetus.
After detection of the ovum (de Graaf 2 nd half 17 century) Malpigy proposed the preformation theory (ovist) which means there is a miniature human inside ovum, that grows after Semen has entered the uterus and grow into a well-developed fetus. This hypothesis was later delegated to spermatozoa. These contradictory and inappropriate beliefs were subject to discussions and dispute, until C.E. Wolf demonstrated that the embryo is a product of the fertilization of ovum by spermatozoa.
800 years prior this the sage Ferdowsi "The Great Iranian Poet" explains nicely the equal participation of man and woman in the production of the fetus and transmission of characters.
After the renaissance and especially in recent years, tremendous achievements have been made in unraveling biological secrets of reproduction. There was no work o n genetics in Iran until 1936, when a genetic course was added to the biology curriculum in related colleges and universities; Iranian Genetics Society was founded in 1966, initiating a steady movement in this field.
Although there was an inevitable gap during the revolution and war in our country, now there is great effort by researchers to eliminate the gap and bring us into the mainstream of world science, and development in biomedical sciences in the third millennium. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| ORIGINAL ARTICLES |
|
|
|
|
An anthropometric and hematological comparison of sickle cell disease children from rural and urban areas |
p. 40 |
HS Nikhar, SU Meshram, GB Shinde
DOI:10.4103/0971-6866.96643 Background: Sickle cell disease (SCD) is a prevalent genetic disorder in India and the rural and urban areas experience distinctly different healthcare facilities. In view of this, a comparative study of SCD-SS pattern children of age 8-15 years from rural and urban areas of Wardha district of Central India was carried out using anthropometric and hematological parameters.
Materials and Methods: The data were collected using standard methods and the results showed a significant (P < 0.05) difference in the mean values for body weight, body mass index (BMI), hemoglobin, hematocrit, and white blood corpuscles (WBC). Statistical analysis of the data was done using SPSS 18.0 software. Individuals were screened by solubility test method. Sickle cell patterns (AS and SS) were determined by using electrophoresis technique.
Result : The SCD-SS children from rural were significantly underweight than those from the urban area of Wardha district. BMI is a good indicator of nutritional status and BMI values of SCD children have less than desired.
Conclusion : The study highlights an urgent need to conduct integrated investigations for SCD population of rural areas covering clinical, nutritional, and social aspects. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
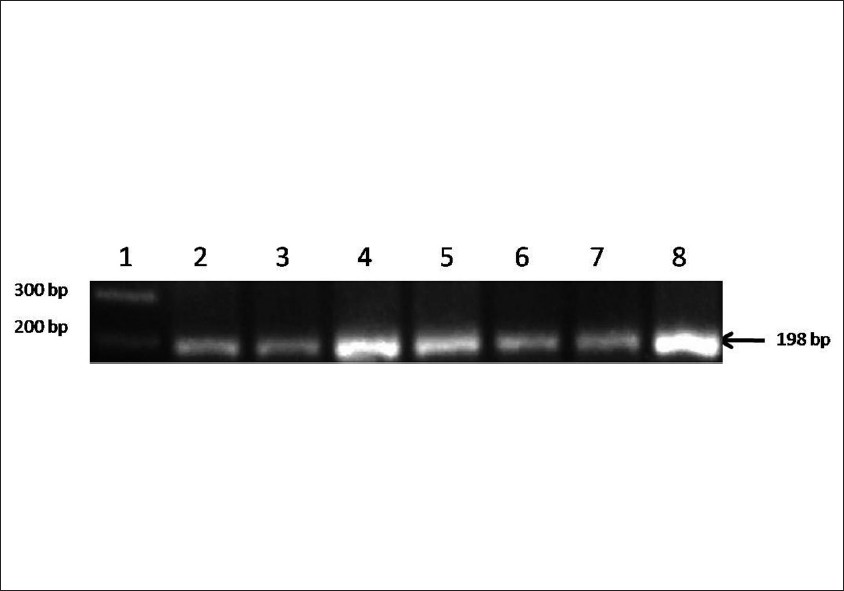 |
Prevalence of methylenetetrahydrofolate reductase C677T polymorphism in eastern Uttar Pradesh |
p. 43 |
Vandana Rai, Upendra Yadav, Pradeep Kumar
DOI:10.4103/0971-6866.96645 Aim: This study was aimed to evaluate the 5, 10-methylenetetrahydrofolate reductase (MTHFR) C677T mutation in eastern Uttar Pradesh population.
Materials and Methods: Polymerase chain reaction (PCR) using specific primers followed by amplicon digestion by Hinf I restriction enzyme was used for MTHFR C677T polymorphism analysis. Total 250 subjects were analyzed.
Results: The CC genotype was found in 192 subjects, followed by CT in 56 subjects and TT in 2 subject. Genotype frequencies of CC, CT and TT were 0.768, 0.224 and 0.008, respectively. The frequency of C allele was found to be 0.88 and that of T allele was 0.12.
Conclusion: It is evident from the results of the present study that the percentage of homozygous genotype (CC) is highest in the target population. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
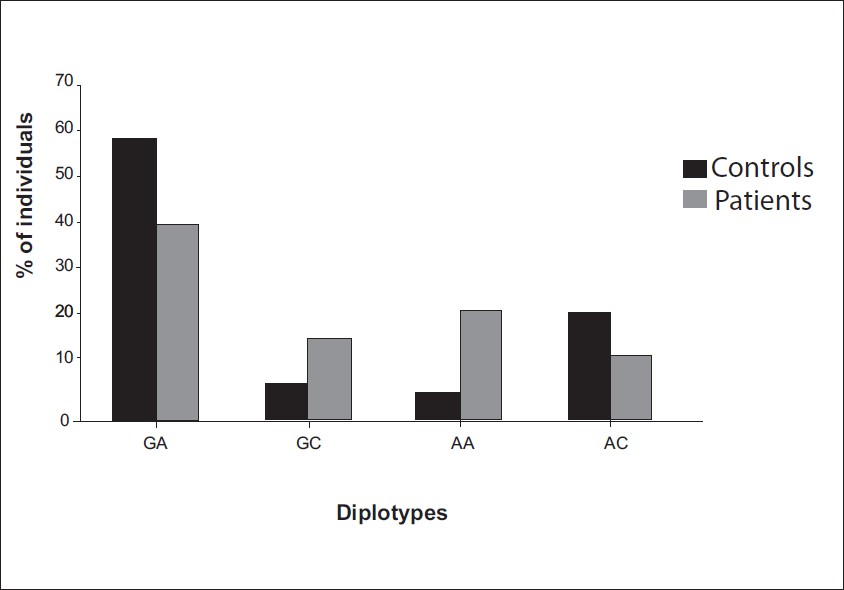 |
Genetic variation in nucleotide excision repair pathway genes influence prostate and bladder cancer susceptibility in North Indian population |
p. 47 |
Rama D Mittal, Raju K Mandal
DOI:10.4103/0971-6866.96648 Background : Inherited polymorphisms of XPD and XPC genes may contribute to subtle variations in NER DNA repair capacity and genetic susceptibility to development of urological cancer such as prostate and bladder cancer.
Materials and Methods: We genotyped four Single Nucleotide Polymorphs (SNPs) of the DNA repair gene XPD and XPC in 195 prostate cancer (PCa) and 212 bladder cancer (BC) patients and 250 healthy controls from the same area. XPD Exon 10 (G>A) by amplification refractory mutation system and Exon 23 (A>C), XPC Intron 9 (Ins/Del) and Exon 15 (A>C) were genotyped by PCR-RFLP.
Results : Variant genotype of XPC demonstrated association with PCa as well as in BC (P, 0.013; P, 0.003). Combined genotype (GA+AA) revealed association with PCa and in BC (P, 0.012, P, 0.002). Variant allele also demonstrated risk in both the cancer. Diplotype of XPD and XPC was associated with a significant increase in PCa and BC risk. Variant (+/+) genotype of XPC intron 9 shown increased risk with PCa and in BC (P, 0.012; P, 0.032). CC genotype of XPC exon 15 revealed increase risk (P, 0.047) with PCa not in BC. In clinopathological grade variant allele of XPC intron 9 and 15 demonstrated risk with high grade of tumor and bone metastasis of PCa. In BC variant allele of XPD exon 10 and 15 also shown association with tumor grade. XPC intron 9 influences the risk of BC in former tobacco users in BC.
Conclusions: Our result support that SNPs in XPD and XPC gene may reduce NER repair capacity and play a major role for PCa and BC in North India. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (13) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Epistatic interactions in idiopathic pulmonary arterial hypertension |
p. 56 |
Shivani Vadapalli, ML Satyanarayana, KL Chaitra, H Surekh Rani, B.K.S. Sastry, Pratibha Nallari
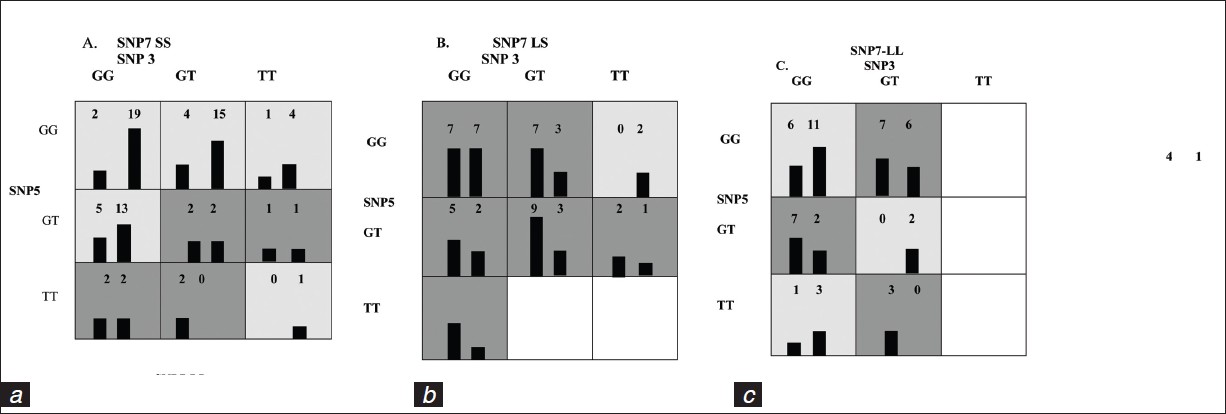
DOI:10.4103/0971-6866.96652 Background : Idiopathic pulmonary arterial hypertension (IPAH) is a poorly understood complex disorder, which results in progressive remodeling of the pulmonary artery that ultimately leads to right ventricular failure. A two-hit hypothesis has been implicated in pathogenesis of IPAH, according to which the vascular abnormalities characteristic of PAH are triggered by the accumulation of genetic and/or environmental insults in an already existing genetic background. The multifactor dimensionality reduction (MDR) analysis is a statistical method used to identify gene-gene interaction or epistasis and gene-environment interactions that are associated with a particular disease. The MDR method collapses high-dimensional genetic data into a single dimension, thus permitting interactions to be detected in relatively small sample sizes.
Aim: To identify and characterize polymorphisms/genes that increases the susceptibility to IPAH using MDR analysis.
Materials and Methods: A total of 77 IPAH patients and 100 controls were genotyped for eight polymorphisms of five genes (5HTT, EDN1, NOS3, ALK-1, and PPAR-γ2). MDR method was adopted to determine gene-gene interactions that increase the risk of IPAH.
Results : With MDR method, the single-locus model of 5HTT (L/S) polymorphism and the combination of 5HTT(L/S), EDN1(K198N), and NOS3(G894T) polymorphisms in the three-locus model were attributed to be the best models for predicting susceptibility to IPAH, with a P value of 0.05.
Conclusion: MDR method can be useful in understanding the role of epistatic and gene-environmental interactions in pathogenesis of IPAH. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Molecular analysis of genetic variation in angiotensin I-converting enzyme identifies no association with sporting ability: First report from Indian population |
p. 62 |
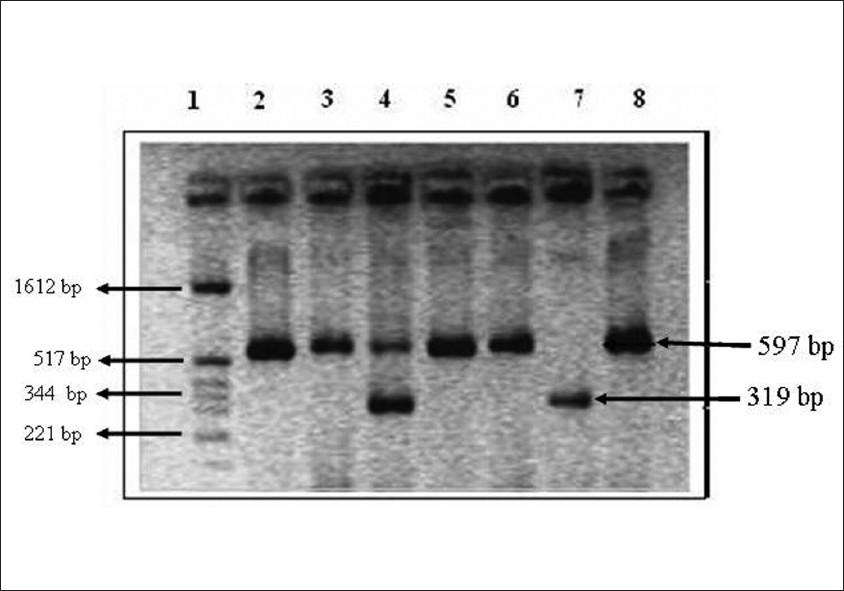
Sweta T Kothari, Pratiksha Chheda, Leena Chatterjee, Bibhu R Das
DOI:10.4103/0971-6866.96653 Introduction: A polymorphism in the angiotensin-converting enzyme (ACE) gene was the first performance enhancing polymorphisms (PEPs) to be identified and correlated with athletic abilities. This polymorphism (rs. 5186) is the absence (deletion; D allele), rather than the presence (insertion, I allele) of 287bp Alu repeat element in intron 16. However, the association of ACE I/D polymorphism in sports abilities have been contradicted and debated. No study has evaluated the ACE gene polymorphism in Indian athletes so far. Hence, the genotype distribution and allelic frequency of ACE gene in selected Indian athletic and non-athletic population was studied.
Materials and Methods: A total of 147 athletes and 131 controls were genotyped for the ACE gene polymorphism using PCR.
Results: No significant association was observed between the allelic frequencies of ACE gene in controls and athletes on a whole, as well as after sub-categorizing the athletes based on the type of sport they played (P > 0.1). However, a higher representation of I allele was observed in the athletes.
Conclusion: ACE genotyping studies need to focus on truly elite athletes of a single sporting discipline, to be able to find an association. The ACE I/D polymorphism may not be considered a marker for human performance, but can be further studied in combination with other potent performance enhancing polymorphisms. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Karyotypic findings in chronic myeloid leukemia cases undergoing treatment |
p. 66 |
Anupam Kaur, Simran Preet Kaur, Amarjit Singh, Jai Rup Singh
DOI:10.4103/0971-6866.96654 Background: Chronic myeloid leukemia (CML) is a clonal myeloproliferative expansion of primitive hematopoietic progenitor cells.
Materials and Methods: In the present study, CML samples were collected from various hospitals in Amritsar, Jalandhar and Ludhiana.
Results: Chromosomal alterations seen in peripheral blood lymphocytes of these treated and untreated cases of CML were satellite associations, double minutes, random loss, gain of C group chromosomes and presence of marker chromosome. No aberrations were observed in control samples. Karyotypic abnormalities have also been noted in the Ph-negative cells of some patients in disease remission.
Conclusion: This is a novel phenomenon whose prognostic implications require thorough and systematic evaluation. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Inbreeding as a cause for deafness: Dadhkai study |
p. 71 |
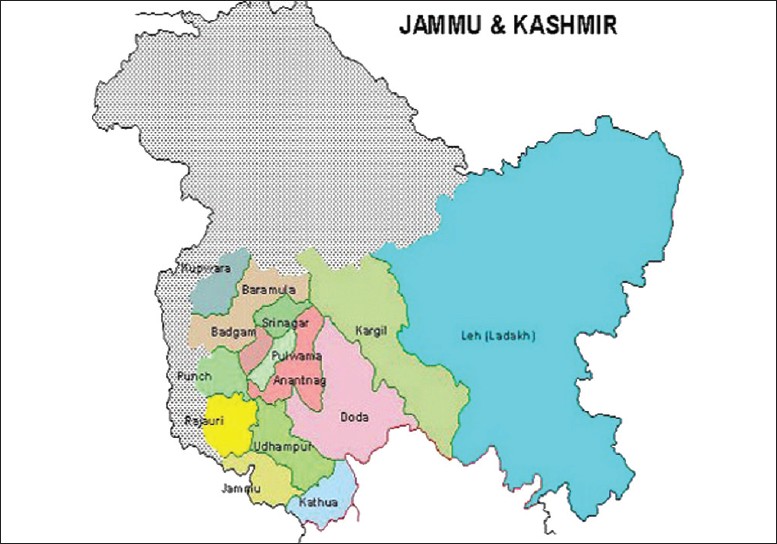
Sushil Razdan, Sunil Kumar Raina, Kamal K Pandita, Shiveta Razdan, Renu Nanda, Rajni Kaul, Sandeep Dogra
DOI:10.4103/0971-6866.96655 Background: We report on the higher prevalence of deaf-mutes from a village in Jammu and Kashmir State of India.
Materials and Methods: A cross-sectional study among 79 deaf mutes using pedigree analysis, audiometry, imaging and molecular analysis.
Results: A high rate of hereditary deafness with 79 individuals diagnosed to be suffering from non-syndrome deafness in a total population of 2452 individuals residing in the village.
Interpretation: Flourishing of intermarriages led to a population with high prevalence of deafness |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Prenatal sonographic evaluation and postnatal outcome of renal anomalies |
p. 75 |
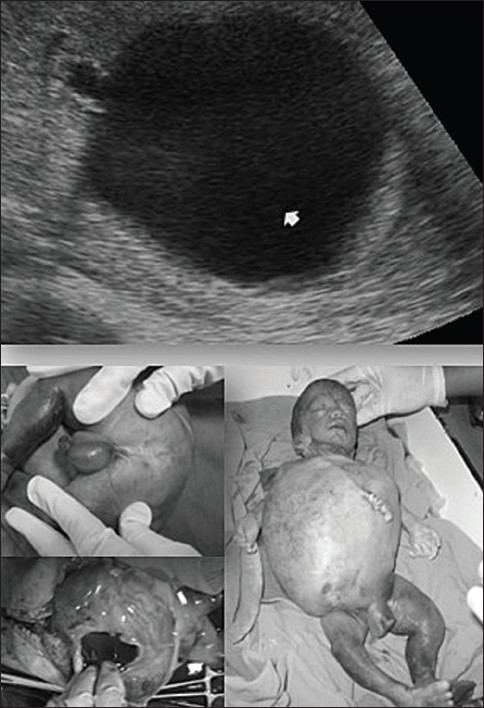
Manisha Kumar, Usha Gupta, Seema Thakur, Shilpi Aggrawal, Jyoti Meena, Sumedha Sharma, Shubha S Trivedi
DOI:10.4103/0971-6866.96656 Objective: To determine the prognosis of antenatally detected renal anomalies by sonographic evaluation.
Materials and Methods: This was a follow-up study of all antenatally detected renal anomalies from January 2008 to Dec 2009 referred to fetal medicine clinic. Prenatal evaluation was done and cases were divided into four groups depending upon their prenatal sonographic findings. Post natal follow-up was done up to one year in cases of live babies. Autopsy was carried out in still born fetus after consent.
Results: The renal anomaly was detected in 55 cases, which were fully followed. The prognosis was said to be poor for group I cases with gross extra renal anomaly along with the renal anomaly, and for group II in which there was organic renal pathology with loss of renal function suggested by non-visualization of bladder and almost absent liquor. Prognosis was guarded and depended upon the gestational age of presentation in group III, which had obstructive uropathy; prognosis was good in group IV cases, which were mild, unilateral or which presented late.
Conclusion: Prenatal sonographic evaluation gives reasonably accurate picture of the prognosis and can be very helpful in counseling the parents regarding prognosis and help in deciding the timing and route of delivery. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
In silico prediction of exposure amino acid sequences of outer inflammatory protein A of Helicobacter pylori for surface display on Eschierchia coli |
p. 83 |
Omid Teymournejad, Ashraf M Mobarez, Zuhair M Hassan, Seyed M Moazzeni, Bagher Yakhchali, Vajihe Eskandari
DOI:10.4103/0971-6866.96659 Background: Outer inflammatory protein A (OipA) is an outer membrane protein of Helicobacter pylori that is involved in inducing IL-8 and intracellular signaling. In this study, we have predicted exposure amino acid sequences of OipA for insertion in permissive sites of CstH subunit of Eschierchia coli CS3 pilli for bacterial surface display.
Materials and Methods: Databases: National Center for Biotechnology Institute and Protein Data Bank. Servers: PHD, SABLE, GOR 4, SignalP3.0, TBBpred, PRODIV-TMHMM, TMRPres2D, CPH Models, PHYRE, GETAREA, VADAR, Pep state and pep window. Software: Swiss PDB viewer and Discovery studio.
Results: In silico prediction of exposure amino acid sequences of OipA led to detection of six sequences of amino acid, 76-87, 106-112, 170-182, 222-230, 242-258, and 278-290. These sequences inserted between amino acid sequences 66-67, 100-101, and 109-110 of CstH that were predicted by Eskandari et al. as permissive sites of CstH.
Conclusion: OipA has the ability to induce IL-8 from gastric epithelial cells and some papers are mentioned that this outer membrane protein involve to attachment and intracellular signaling. Receptor of OipA and adhesion motifs on this protein is unknown. Detection of exposure motifs aids to recognition of adhesion motifs and receptor of OipA on gastric epithelial cells. In this study, we have predicted exposure amino acid sequences for insert to subunit CstH of CS3 pilli E. coli for surface display. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
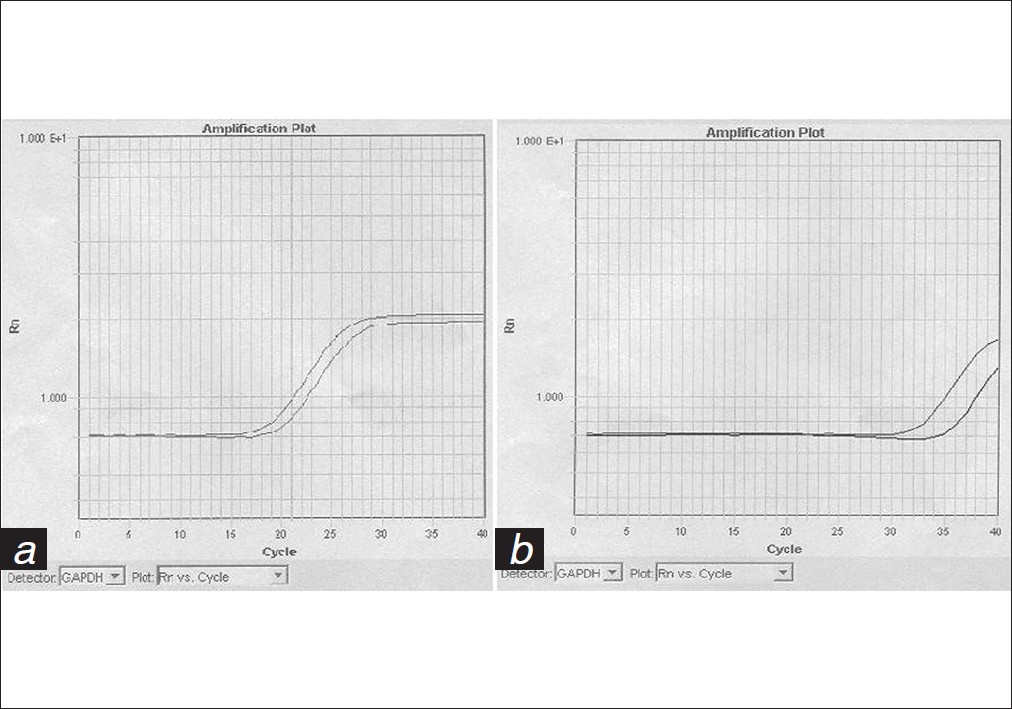 |
SRY sequence in maternal plasma: Implications for non-invasive prenatal diagnosis: First report from India |
p. 87 |
Edna D'Souza, Sona Nair, Anita Nadkarni, Kanjaksha Ghosh, Roshan B Colah
DOI:10.4103/0971-6866.96661 Aim: The presence of circulatory cell-free fetal DNA in maternal plasma has found new applications in non-invasive risk-free prenatal diagnosis.
Materials and Methods: We made use of a size separation approach along with real time polymerase chain reaction (PCR) to evaluate the use of fetal DNA in the detection of the sex of the fetus. Cell-free fetal DNA was isolated from the plasma of 30 women (10-20 weeks gestation) using a size separation approach. We made use of Taq Man Chemistry and real time PCR using primers and probes for GAPDH and SRY.
Results: Only 24 cases could be studied as there was no amplification in six cases. Fetal sex was accurately determined in all of the 24 cases wherein 19 women were carrying male fetuses and five women were carrying female fetuses. An increase in the amount of fetal DNA was observed with an increase in the gestational age.
Conclusions: Real time PCR analysis is a highly sensitive and accurate tool for non-invasive prenatal diagnosis, allowing detection of the sex of the fetus as early as 10 weeks of gestation. Non-invasive prenatal diagnosis eliminates the risk of fetal loss associated with the invasive procedure. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Sword Plugin for Repository]Beta |
|
|
|
|
|
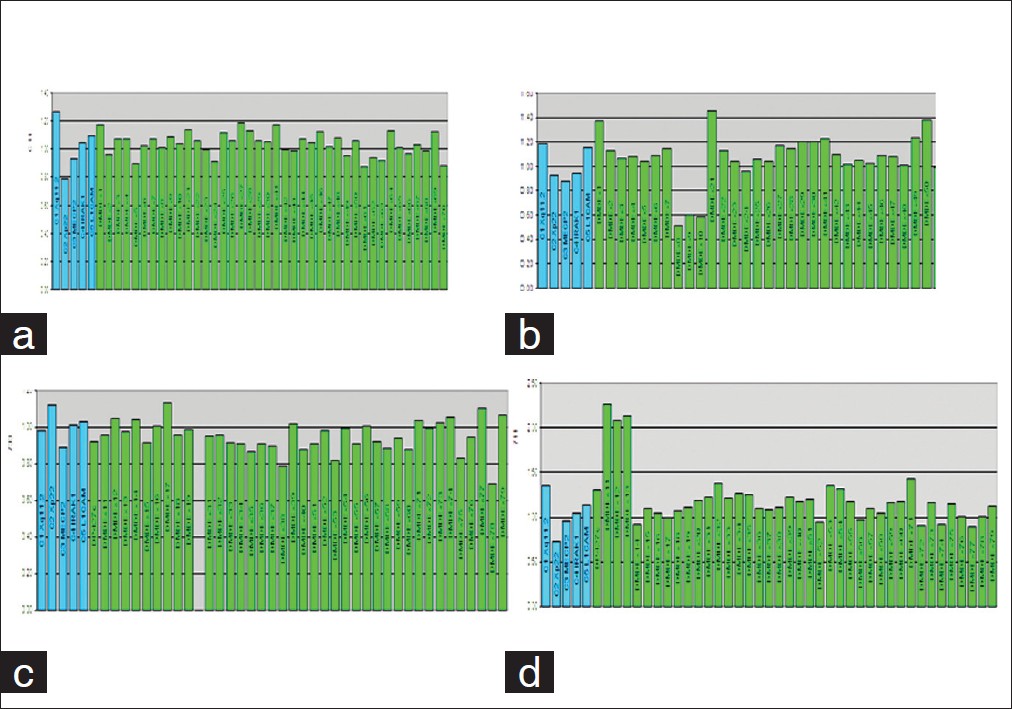 |
Utility of MLPA in mutation analysis and carrier detection for Duchenne muscular dystrophy |
p. 91 |
Prashant K Verma, Ashwin Dalal, Balraj Mittal, Shubha R Phadke
DOI:10.4103/0971-6866.96667 Context: Multiplex ligation probe amplification (MLPA) is a new technique to identify deletions and duplications and can evaluate all 79 exons in dystrophin gene in patients with Duchenne muscular dystrophy (DMD). Being semi-quantitative, MLPA is also effective in detecting duplications and carrier testing of females; both of which cannot be done using multiplex PCR. It has found applications in diagnostics of many genetic disorders.
Aim: To study the utility of MLPA in diagnosis and carrier detection for DMD.
Materials and Methods: Mutation analysis and carrier detection was done by multiplex PCR and MLPA and the results were compared.
Results and Conclusions: We present data showing utility of MLPA in identifying mutations in cases with DMD/BMD. In the present study using MLPA, we identified mutations in additional 5.6% cases of DMD in whom multiplex PCR was not able to detect intragenic deletions. In addition, MLPA also correctly confirmed carrier status of two obligate carriers and revealed carrier status in 6 of 8 mothers of sporadic cases. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
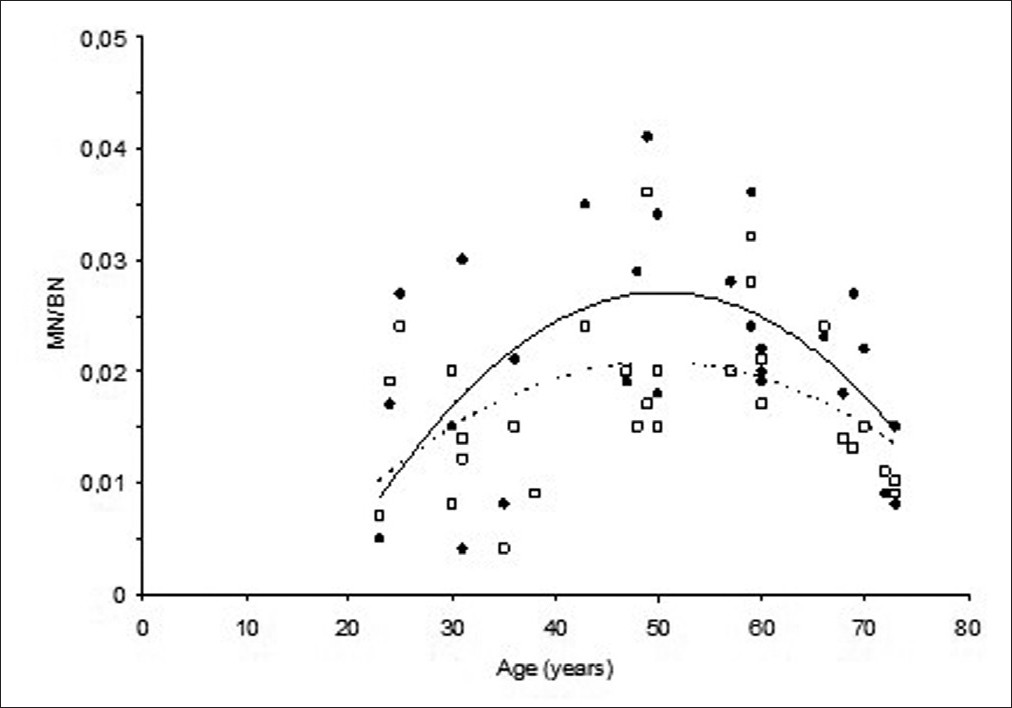 |
The effect of aging on micronuclei frequency and proliferation in human peripheral blood lymphocytes |
p. 95 |
Tuncay Orta, Süreyya Günebakan
DOI:10.4103/0971-6866.96671 Introduction: Increase in the instability of cellular genome with an increasing age is the result of an accumulation of cellular damage and mutations. This instability which might be observed as chromosome damage or chromosome losses can be measured by the micronucleus technique.
Aim: The aim of this study was to investigate the effect of aging and oxidative stress induced by non-toxic levels of H 2 O 2 on micronuclei induction and their relationship to cell proliferation in human peripheral blood lymphocytes.
Materials and Methods: Healthy volunteers with different ages were choosen. Spontaneous and H 2 O 2 induced micronuclei frequencies were measured in peripheral blood lymphocytes of 30 volunteers by the micronucleus method.
Results: Spontaneous micronuclei frequencies increased first then started to decrease after 50 years of age. This biphasic response was significantly higher than micronucleus (MN) frequencies induced by H 2 O 2 (P < 0.05), which followed the similar shape of response to increasing ages with lower frequencies. Proliferative capacity of cells either treated with H 2 O 2 or not did not differ with an increasing age giving similar responses.
Conclusion: These results indicate biphasic character of chromosome damage; first increase and decrease after 50 years with an increasing age. But this change pattern was not correlated with the steady state of proliferation capacity of cells through an increasing age. Decreases in H 2 O 2 -induced MN frequencies compared to spontaneous MN frequencies may be inducing an apoptosis by H 2 O 2 treatment leading to underscoring damaged cells. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (4) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
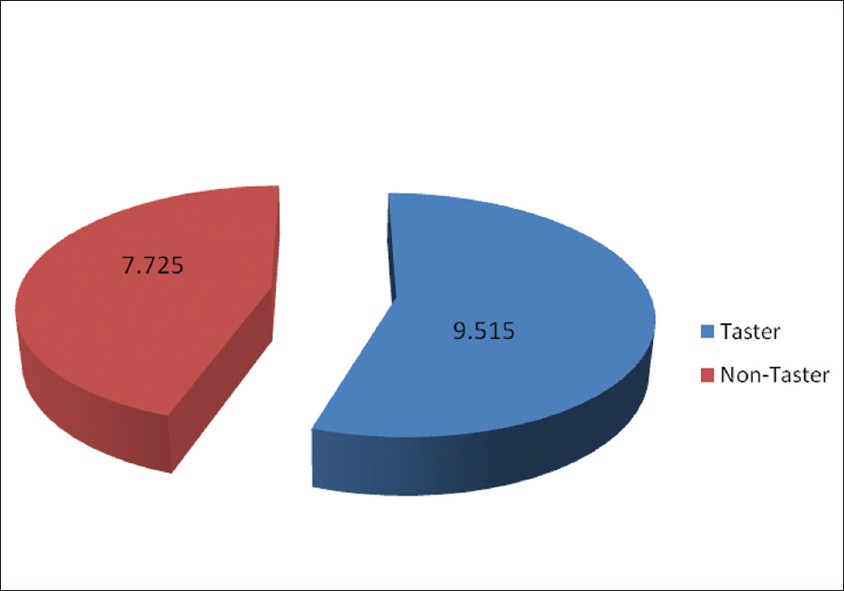 |
Genetic sensitivity to bitter taste of 6-n Propylthiouracil: A useful diagnostic aid to detect early childhood caries in pre-school children |
p. 101 |
Raghavendra Pidamale, B Sowmya, Ann Thomas, Tony Jose
DOI:10.4103/0971-6866.96672 Purpose: Genetic factor to bitter taste perception appears to be largely mediated by the TAS2R38 gene. The insensitivity to bitter compounds like 6-n-propylthiouracil (PROP) is mediated by this gene. PROP, a pharmacological drug used in treatment of Graves' disease, proved to be useful tool in determining the genetic sensitivity levels to bitter and sweet taste. The purpose of this study is to show much simpler PROP sensitivity technique for the clinical examiner and its application as a diagnostic aid in Early Childhood Caries (ECC) detection among preschool children.
Materials and Methods: A total of 119 children belonging to the age group of 36 to 71 months of both sexes, were recruited from A. J. Institute of Dental Sciences, Mangalore (Karnataka). PROP sensitivity test was carried out to determine the inherent genetic ability to taste a bitter or sweet substance. This study used simpler scaling method to find out genetic sensitivity to bitter taste; one who tasted bitter as taster and one who was not able to differentiate/tasted like paper as non-taster. A questionnaire was provided to evaluate their dietary habits and caries experience was recorded. Collected data were tabulated and subjected to statistical analysis.
Results: In the total of 119 children the mean dmfs was definitely higher in non-taster children compared to tasters. The tasters had a mean dmfs value of 9.5120 (S.D. 7.0543) and non-tasters had a value of 7.7250 (S.D. 8.33147), which was statistically significant. The results suggested that there was increase in caries experience among the group of non-tasters as compared to tasters. Tasters tended to be sweet dislikers and non-tasters tended to be sweet likers. On the whole, tasters had a bad dentition as compared to non tasters.
Conclusion: The PROP sensitivity test (filter paper test) proved to be a useful diagnostic tool in determining the genetic sensitivity levels of bitter taste. The knowledge of a child's taste perception can help us in identifying the children who are at higher risk for ECC. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (3) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| BRIEF REPORTS |
|
|
|
|
Trisomy 8 in leukemia: A GCRI experience |
p. 106 |
Sonal R Bakshi, Manisha M Brahmbhatt, Pina J Trivedi, Esha N Dalal, Dharmesh M Patel, Sejal S Purani, Shilin N Shukla, Pankaj M Shah, Prabhudas S Patel
DOI:10.4103/0971-6866.96673 Trisomy of chromosome 8 is frequently reported in myeloid lineage disorders and also detected in lymphoid neoplasms as well as solid tumors suggesting its role in neoplastic progression in general. It is likely to be a disease-modulating secondary event with underlying cryptic aberrations as it has been frequently reported in addition to known abnormalities contributing to clinical heterogeneity and modifying prognosis. Here, we share our findings of trisomy 8 in leukemia patients referred for diagnostic and prognostic cytogenetic assessment. Total 60 cases of trisomy 8, as a sole anomaly or in addition to other chromosomal aberrations, were reported (January 2005-September 2008). Unstimulated bone marrow or blood samples were cultured, followed by GTG banding and karyotyping as per the ISCN 2005. Patients with +8 were chronic myeloid leukemia (CML) (36), acute myeloid leukemia (AML) (17), and acute lymphoblastic leukemia (ALL) (7). In 7 patients, trisomy 8 was the sole anomaly, whereas in 6 patients +8 was in addition to normal clone, in 47 patients, the +8 was in addition to t(9;22), t(15;17), and others, including 3 with tetrasomy 8. Only one patient showed constitutional +8. The present study will form the basis of further cumulative studies to correlate potential differential effects of various karyotypic anomalies on disease progression and survival following a therapeutic regime. To unravel the role of extra 8 chromosome, constitutional chromosomal analysis and uniparental disomy will be considered. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Recombination in human leukocyte antigen region in two Asian Indian families |
p. 109 |
Mahendra N Mishra, Ajay Sharma
DOI:10.4103/0971-6866.96674 Background: Recombination (crossing over) may generate novel haplotypes that can be beneficial to a population against recently introduced pathogens. It may lead to the generation of new alleles.
Settings and Design: A prospective study at a tertiary care centre.
Aim :0 To report two rare cases of crossing over in HLA region.
Materials and Methods: Tissue-typing was done by sequence specific primers (SSP) for DR locus and by both SSP and serology for Class I which was reconfirmed on fresh samples.
Results: In one patient crossing over had taken place in the region of A locus resulting in inheritance of A*01 instead of expected A*11. In second family crossing over had taken place in region of DRB1 locus and the sibling inherited DRB1*08 instead of DRB1*10.
Conclusions: Possibility of recombination must be considered when interpreting implausible tissue-typing results of families worked up for BMT. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| CASE REPORTS |
|
|
|
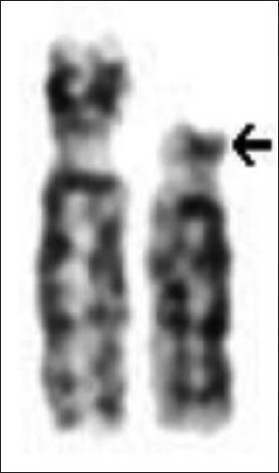
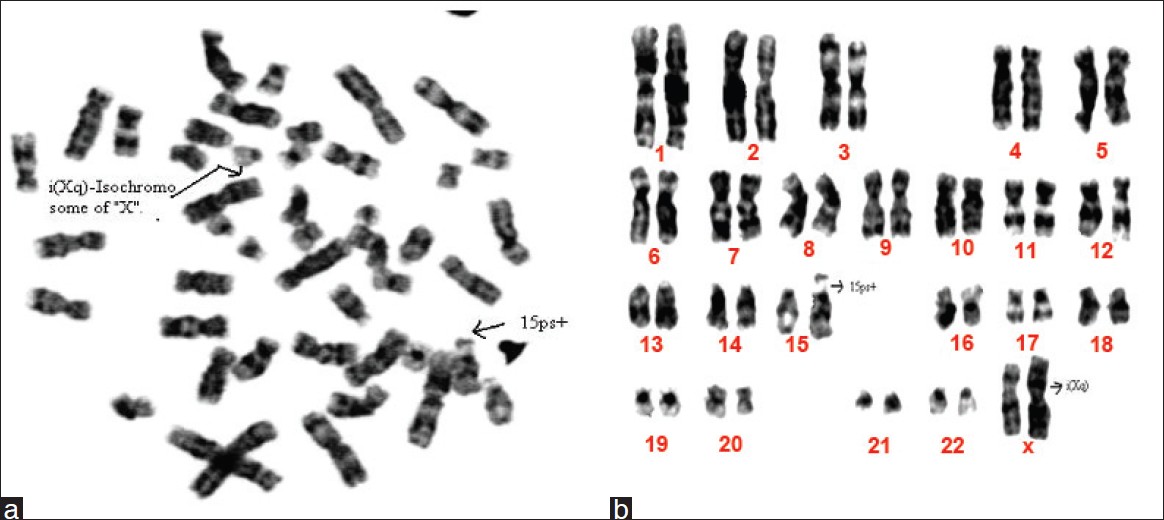 |
Cytogenetic investigation of patients with primary amenorrhea |
p. 112 |
K Vijaya Laxmi, SJ Babu, S Dayakar, RN Mehrothra, Kalal I Goud
DOI:10.4103/0971-6866.96676 Primary amenorrhea refers to absence of spontaneous menarche even after the age of 16. Cytogenetic analysis in two cases with primary amenorrhea, short stature, poorly developed secondary sexual characteristics, and growth retardation were studied. Routine GTG-band analysis of metaphases from peripheral blood leucocytes revealed female karyotype with a 15(ps+) and an isochromosome of X, i(Xq), in one patient and 46,X, i(Xq), in another patient. Ascertainment of the karyotype aided in confirmation of the provisional diagnosis, a better phenotype-genotype correlation to understand clinical heterogeneity in genetic counseling. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Wolf-Hirschhorn syndrome: A case demonstrated by a cytogenetic study |
p. 117 |
Yamini S Pokale, Ajinkya M Jadhav, Ushang Kate
DOI:10.4103/0971-6866.96677 We present a case with a 4p terminal deletion, evidenced in GTG-banded chromosome study. Phenotypic signs described in the classical Wolf-Hirschhorn syndrome were found on clinical examination of our patient. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Familial germ cell tumor |
p. 119 |
Sanju Cyriac, Rejeev Rajendranath, A Robert Louis, TG Sagar
DOI:10.4103/0971-6866.96679 Familial testicular germ cell tumors are well known in literature. Only few cases are reported where both brother and sister of the same family suffered from germ cell malignancies. We present a family where the proband is a survivor of ovarian dysgerminoma stage IA. Her elder male sibling became acutely ill and was detected to have disseminated testicular malignancy with grossly elevated markers and vegetations in the mitral valve leaflets. Despite all measures he could not be saved. Presence of germ cell malignancies in the siblings of different sex in the same family points toward a genetic susceptibility. Literature review revealed only six similar cases. A discussion regarding the rare occurrence of familial germ cell malignancies with the affected family members may be worthwhile. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
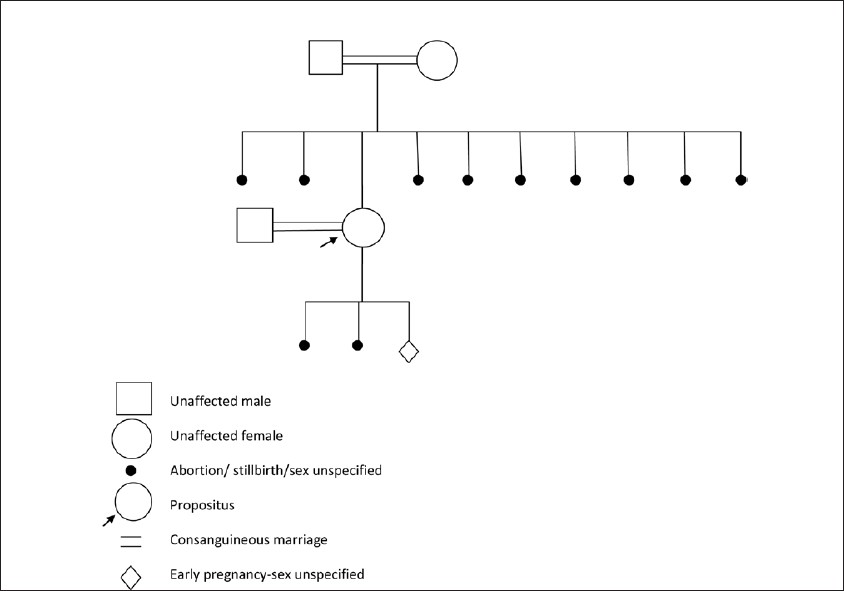 |
Methylenetetrahydrofolate reductase mutations, a genetic cause for familial recurrent neural tube defects |
p. 122 |
Laxmi V Yaliwal, Rathnamala M Desai
DOI:10.4103/0971-6866.96680 Methylenetetrahydrofolate reductase (MTHFR) gene mutations have been implicated as risk factors for neural tube defects (NTDs). The best-characterized MTHFR genetic mutation 677C→T is associated with a 2-4 fold increased risk of NTD if patient is homozygous for this mutation. This risk factor is modulated by folate levels in the body. A second mutation in the MTHFR gene is an A→C transition at position 1298. The 1298A→C mutation is also a risk factor for NTD, but with a smaller relative risk than 677C→T mutation. Under conditions of low folate intake or high folate requirements, such as pregnancy, this mutation could become of clinical importance. We present a case report with MTHFR genetic mutation, who presented with recurrent familial pregnancy losses due to anencephaly/NTDs. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
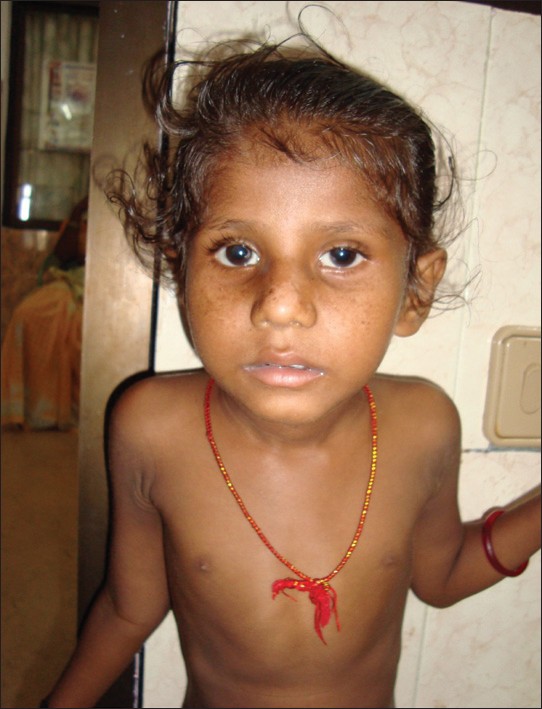 |
Cockayne syndrome-xeroderma pigmentosum complex with demyelination: A rare association |
p. 125 |
Usha Rani Singh, Shujaath Asif, Peter Prasanth Kumar Kommu, Philomina D'Souza
DOI:10.4103/0971-6866.96681 Xeroderma pigmentosum-Cockayne syndrome (XP-CS) includes facial freckling and early skin cancers typical of XP and some features typical of CS, such as mental retardation, spasticity, short stature, and hypogonadism. XP-CS does not include skeletal involvement, the facial phenotype of CS, or CNS demyelination and calcifications. We present a rare patient whose genome probably harbored a specific combination of mutations producing a rare double syndrome of XP-CS, with facial phenotype of CS, and CNS demyelination. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Sword Plugin for Repository]Beta |
|
|
|
|
|
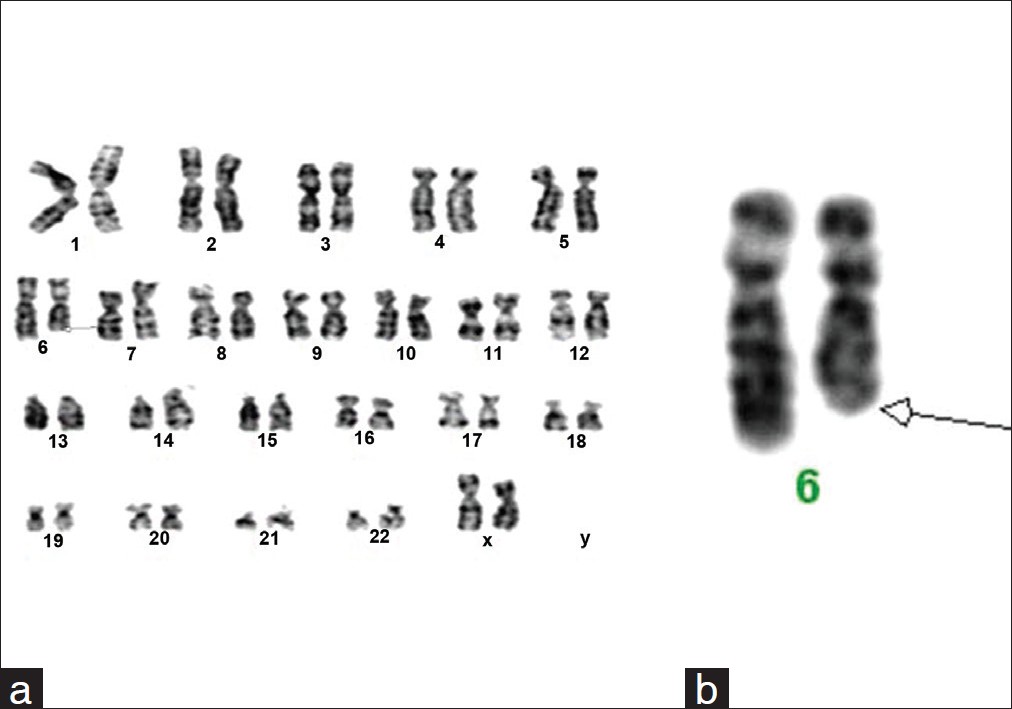 |
Dysmorphic features and congenital heart disease in chromosome 6q deletion: A short report |
p. 127 |
Sreelata Nair, Rini Varghese, Sajeed Hashim, Pappachan Scariah
DOI:10.4103/0971-6866.96682 In this report, we describe a one and a half year old girl showing terminal deletion of long arm of chromosome 6q. The associated abnormalities such as congenital heart disease, mental retardation, and dysmorphic features are described. Cytogenetic studies with GTG banding showed 46,XX,del(6)(q24→qter). Karyotype of the parents was normal suggesting a denovo event. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
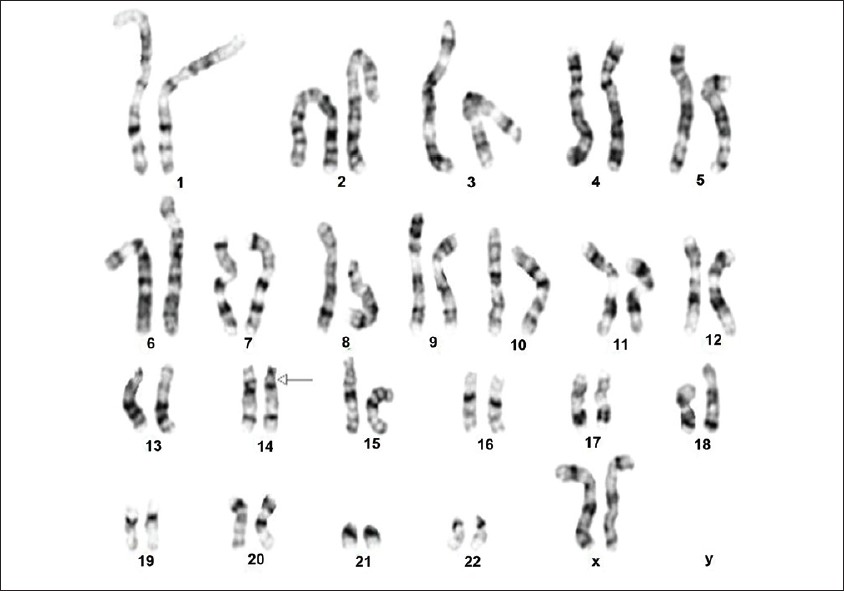 |
A child with mosaicism for deletion (14)(q11.2q13) |
p. 130 |
Thilini H Gamage, Imaya U.H. Godapitiya, Shakila Nanayakkara, Rohan W Jayasekara, Vajira H.W. Dissanayake
DOI:10.4103/0971-6866.96684 In this case report we describe a child with a de novo deletion in the (q11.2q13) region of chromosome 14. The child presented with dysmorphic features - anophthalmia, microcephaly, and growth retardation. Cytogenetic studies showed mosaicism. The karyotype was 46,XX,del(14)(q11.2;q13) [16] /46,XX [9]. We compared the features observed in this child with that of others with the same deletion reported in scientific literature and found that this is the first report of a child mosaic for this deletion. It is also the first time it has been reported in association with anophthalmia. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
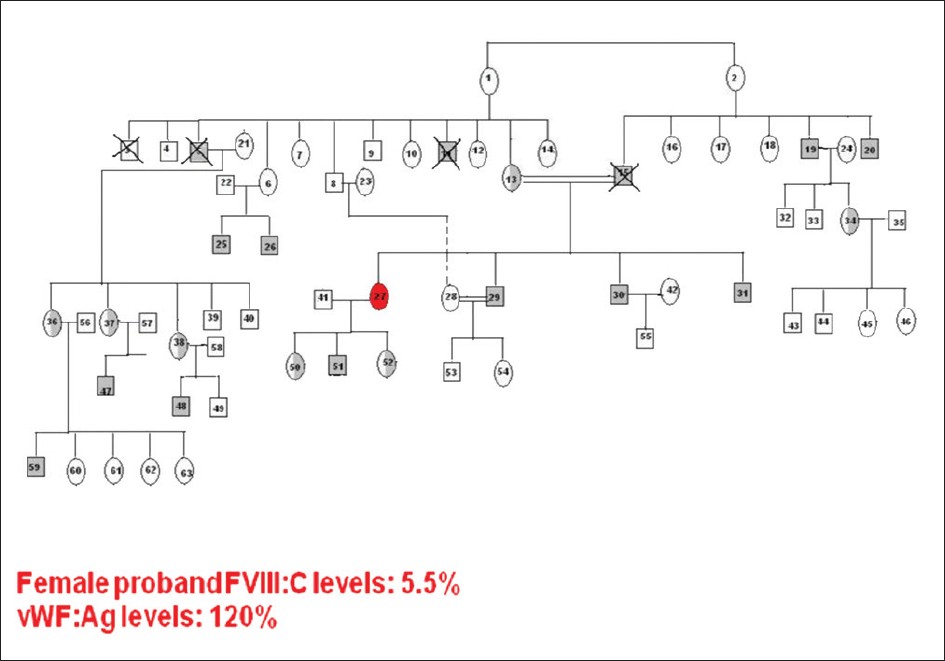 |
A homozygous female hemophilia A |
p. 134 |
Preethi S Nair, S Shetty, Kanjaksha Ghosh
DOI:10.4103/0971-6866.96685 Background: Hemophilia A (HA), being an X-linked recessive disorder, females are rarely affected, although they can be carriers.
Aims: To study the mutation in F8 gene in an extended family with a homozygous female HA.
Materials and Methods: All the seven affected members (six males and one female) were initially screened by Conformation Sensitive Gel Electrophoresis (CSGE) and direct DNA sequencing.
Results: A homozygous missense mutation c.1315G>A (p.Gly420Ser) was identified in exon 9 of F8 gene in homozygous state in the affected female born of 1Ί consanguinous marriage and in all the affected male members of the family. Her factor VIII levels was found to be 5.5%, vWF:Ag 120%.
Conclusion: In India, as consanguineous marriages are very common in certain communities (up to 30%), the likelihood of encountering female hemophilia is higher, although this is the first case of HA out of 1600 hemophilia families registered in our Comprehensive Haemophilia Care Center. Genetic diagnosis in such cases is not necessary as all the male children will be affected and daughters obligatory carriers. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| PERSONAL REFLECTION |
|
|
|
|
A comparison of Fulani and Nadar HLA |
p. 137 |
Clyde Winters
DOI:10.4103/0971-6866.96686 Here recent studies of Nadar and Fulani HLA-A and HLA-B were compared to determine if these populations were related. The analysis revealed that the Nadar and Fulani populations share a number of unique alleles including A*101, A*0211, A*03011, A*3303, B*3501, B*3701, and B*51011. The study suggests a former residence of these diverse populations in same geographical area. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Sword Plugin for Repository]Beta |
|
|
|
|
|










